A review of xenobiotic acyl CoA thioester formation
Abstract
From the time unknown, invention and innovation have been vital for human development. In the healthcare sector, pharmaceutical companies have worked extensively on developing new entities that have become therapeutic for diseases. However, these companies face problems with poor revenue generation; without revenue, they cannot develop new drugs. The problems are mainly due to the delay in acquiring FDA approval, the withdrawal of drugs or their rejection as a result of post- marketing surveillance. Several drugs, mostly acidic, have been discontinued due to the occurrence of idiosyncratic drug reactions (IDRs) in past decades. IDRs are troublesome as they are mainly identified after the drug has been launched in the market. It is very difficult to predict their occurrence pre-clinically. However, the metabolism of carboxylic acid is presumed to be a potential contributor. Two pathways involved in carboxylic acid metabolism may lead to chemically reactive metabolites: (I) Acyl glucuronide formation and (II) Acyl-CoA thioester formation. In several studies there has been evidence of toxicity due to glucuronides in most of the drugs, whereas little information has been documented about thioesters. The high reactivity and involvement of xenobiotic thioesters in crucial biological pathways such as β-oxidation etc. would make it interesting to investigate their disposition. Evidently, their involvement in toxicity is significant and requires exploration; xenobiotic thioesters should be handled as ‘structural alerts’ in causing adverse drug reactions (ADRs).
1.0 Drug failure and economical consequences
In the healthcare sector, pharmaceutical companies have worked extensively on developing new entities that are therapeutic for diseases. However, poor revenue is still a problem, mostly due to the withdrawal of drugs or their rejection on the basis of post-marketing surveillance (1). Different surveys have estimated that between 1970 and 1982, the approximate cost for launching a drug in the market was US$114 million (2), in 2003 it was US$802 million (3) and in 2010, US$1 billion (4).
![Figure 1. Decrease in the approval rate of drugs from 1996 to 2009 [5]](https://www.ivoryresearch.com/wp-content/uploads/2013/04/kate-griffin-fig-1.jpg)
Figure 1. Decrease in the approval rate of drugs from 1996 to 2009 [5]
In 2000, 30% drugs were withdrawn due to poor drug safety (6). In 2010, the total estimated revenue loss to the companies due to the discontinuation of drugs was approximately US$74 billion (7). The root causes of attrition are lack of efficacy, toxicology and clinical safety. Suitable measures need to be implemented to eliminate the percentage of drug failure. For instance, providing strong evidence to show proof of mechanism and proof of concept, identification of toxic metabolites and the use of appropriate animal models which effectively mimic the effect of the drug in humans could be useful (1).
2.0 Adverse drug reactions (ADRs)
The concept of toxicity is not new; it was known in the time of the Greeks (8). Paracelsus differentiated between drugs and poison by dose level and postulated that no drug is safe at all doses (9). The World Health Organization (WHO) defines ADRs as ‘any noxious, unintended and undesired effect of a drug, which occurs at doses used in humans for prophylaxis, diagnosis, or therapy’ (10). ADRs are categorised as a major cause of hospitalisations (11-13). A French study shows that in the 1970s, 1.8% cases of total hospital admissions were due to drug toxicity (14). Very little is known about drug safety as new drugs have been, on the average, exposed to only about 1500 patients before getting the license (15). Investigations of incidences of ADRs between 1966 and 1996 ranked them as the fourth to sixth leading cause of death in the U.S (16). A recent survey in Sweden indicates that ADRs are the seventh leading cause of morbidity and mortality and shows that the most frequently involved drugs are antithrombotic drugs, non-steroidal anti-inflammatory drugs (NSAIDs), antidepressants and cardiovascular drugs (17). Of the 29 drugs withdrawn in the UK, the U.S. and Spain between 1974 and 1993, nine of them were classified as NSAIDs (18). ADRs become troublesome when they are not detected pre-clinically but are noticed in the population after the drug has been launched in the market. The cause of such reactions is thought to be by direct or indirect pathways (19). However, a basic understanding of the occurrence of these events has not been established. Further research is required in this area to outweigh benefits over the risks involved in a drug development.
| Type A | Type B | Type C | Type D |
| Predictable, dose-related, depends on the pharmacology of drug. | Causes iatrogenic diseases, are dose-independent and are unpredictable. | Predictable. These reactions are associated with long-term therapies. | These are delayed reactions like carcinogenicity and teratogenicity. |
2.1 Type B or idiosyncratic drug reactions (IDRs)
IDRs are seen only at later phases of discovery and are associated with public health problems. They mostly occur after the drugs have entered the market, thus causing a heavy financial loss to the pharmaceutical industry (21). The reactions to the drugs are uncommon and bizarre and are known to contribute 25% to ADRs (16). They mostly they cause problems to the liver, as the liver is intensively involved in metabolism, detoxification and oxidation reactions (22-23). More than 600 drugs have been associated with hepatotoxicity over the years (24) with blood dyscracias, skin toxicities and kidney and nervous system disorders being exhibited(20, 25). Between 1975 and 1999, approximately 10% of the drugs launched in the market were given a ‘black-box’ warning by the FDA or were taken off the market due to reported incidences of IDRs (26).
The majority of the drugs shown to cause toxicity belong to the class of NSAIDs (27), anti-epileptic drugs (28) and lipid lowering agents (29-30). In 1998, the FDA received reports of 68 deaths caused by Bromfenac (Duract), 17 of which were liver injuries (31). Most recently, Lumiracoxib, an analogue of Diclofenac, COX-2, a selective NSAID, was taken off the market after reports of serious liver injuries (32). Salicylic acid, which contains the drug aspirin, has been associated with gastrointestinal toxicity (33). In the past decades, many NSAIDs have been correlated with gastrointestinal problems, liver injuries and dyspepsia (34- 47).
2.1.2 Idiosyncrasies- a serious problem
The reasons for these IDRs remains elusive as the symptoms are not seen in the entire population, but are idiosyncratic; i.e. they remain restricted to certain sub-groups. In addition, the availability of suitable animal models that can be used to predict their occurrence is in a nascent phase, making it more difficult to detect problems at earlier stages. It has been proposed that one of the contributory factors to susceptibility to IDRs is drug metabolism. It is known that metabolism can exhibit marked species variability and inter-individual variability, depending on the ethnic group, gender, genotype and age (48-52).
Metabolic reactions are known to inactivate xenobiotic compounds by making them soluble to allow easy excretion by the kidneys. However, metabolism may also lead to the production of chemically reactive species, which can be toxic. Interestingly, carboxylic acid is present in all NSAIDs that have been withdrawn from the market (See Section 2.1). Based on this finding, the presence of carboxylic acid is thought to be one of the reasons for toxicity. However, the reasons for assuming that carboxylic acid is one of the causes of hepatotoxicity are still limited, indirect and based on the knowledge that carboxylic acid is metabolised to produce reactive intermediates, most notably toxic. Thorough exploration of the metabolism of acidic drugs is required to combat or predict the occurrence of toxicities
3.0 Metabolism of carboxylic acid
Metabolism has two phases; Phase I and Phase II. Phase I involves basic enzyme- catalyzed reactions such as oxidation, hydrolysis and reduction. After every reaction, the test compound contains a chemically reactive group like –OH, -COOH and -NH2 that serves as a reactive chemical moiety for phase II enzymes, for conjugation reaction (53). Later, the body excretes the metabolised drug.
Drugs could elicit pharmacological effects either by directly acting on the body or by forming chemically reactive metabolites as a part of their metabolism. (54-55). The modification of drug moiety into its metabolites is termed biotransformation (56).
Metabolism of carboxylic acid occurs via two major pathways – UDP-Glucuronosyl-tranferase (UGT) catalysed in conjugation with glucuronic acid and acyl coenzyme A synthetase/ligase (CoASH) catalysed formation of acyl CoA thioester (Fig. 4). The prevalence of a particular pathway depends on factors like substrate affinity and co-factor availability (55). Glucuronide formation takes priority at high concentrations (57, 58). Both glucuronides and thioesters are electrophilic and could elicit toxicity, if they escape inactivation by S- glutathione thioester formation as a part of the Phase II conjugation reaction (55).
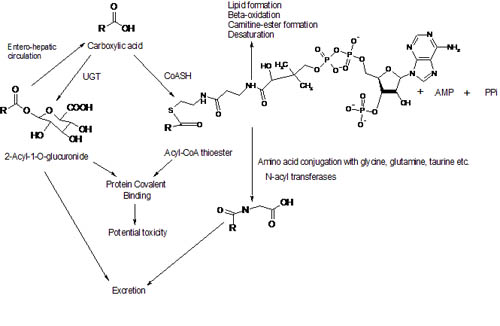
Figure 4 Overview of carboxylic acid metabolism and some processes in which the metabolites may be involved
3.1 Glucuronidation
Glucuronidation has been widely explored as a major pathway in carboxylic acid metabolism (59-60). It yields polar β-1-O-Acyl glucuronides by utilising UGTs (61). They are released into bile by ATP-dependent specific conjugate export pumps including multi-drug resistant associated proteins (MRP2) where they undergo β- glucuronidase mediated hydrolysis to form aglycone. The Hydroxyl group on a glucuronic ring can undergo intermolecular-Acyl migration to yield β-glucuronidase resistant isomers (60). Glucuronides can covalently bind to proteins and form drug-protein adducts which could behave as antigens and instigate an immune response in the body as proposed by the ‘Hapten hypothesis’ (43). Glucuronides are also known to accumulate in the bile and cause insufficient bile secretion or be transported to the intestine and cause ulcerations and erosions (62). Clofibrate and gemofibrozil acyl glucuronide can form DNA adducts and cause genotoxicity (55). The formation of glucuronides varies in species and between individuals and therefore, it has been speculated, may be a potential candidate for idiosyncratic drug reactions (63-64). One of the most studied drugs for glucuronide toxicity is Diclofenac. Although it is still on market, it has been associated with number of drug-induced toxicities (65-69). Drug- protein adducts formation by acyl glucuronides has also been evidenced by the use of NSAIDs like Zomepirac (70-71), Benoxaprofen and Flunoxaprofen (72).
3.2 Acyl- CoA thioester formation
The biochemistry of the formation of Acyl CoA thioesters is elucidated, based on the understanding of lipid metabolism. Acyl CoA thioester formation is catalysed by CoASH in the presence of ATP (Fig. 4) (61).
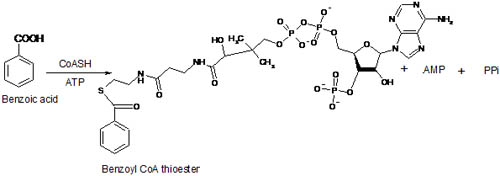
Figure 5. Formation of Benzoyl CoA thioester
Initially, it was thought that only one enzyme is involved in acid metabolism. However, isolation of Salicyl CoA (73) negated that assumption and several studies have deduced that there is an involvement of many isomers of CoA ligase (74-76). CoASH is classified according to substrate specificity and cellular location into three families; short chain, medium chain and long chain (61). This suggests its broad substrate spectrum and, therefore, its inability to effectively differentiate between endogenous and exogenous fatty acids. The evidence that strongly supports this hypothesis is the competitive behavior of (R)-ibuprofen with endogenous palmitic acid, which both require long chain fatty acyl synthetase for metabolism (77).
Recent examples of detection and identification of acyl CoA thioesters include Zomepirac, Naproxen, Clofibric acid, Tolmetin and Ibuprofen, which are associated with toxicities (78-83).
4.0 Thioester associated toxicity
To date, there has been compelling evidences to suggest that acyl glucuronides are xenobiotic toxic derivatives in the body but very limited data is available for Acyl CoA thioesters involvement. However, the number of metabolic pathways (Fig.4) in which acyl-CoAs participate suggest that it would be interesting to investigate their role in toxicity. The following is the discussion about events related to the formation of Acyl CoA thioesters which could be involved in toxicity.
4.1 Reactivity and conjugation reaction
Formation of Acyl CoAs increases the reactivity of carboxylic acid by acidifying proton at α and β carbon (84). Thioesters, like glucuronides, can covalently bind to proteins and form drug-protein adducts (79). One of the earliest studies on chemical reactivity of Acyl CoAs was demonstrated in Salicyl-CoA, which exhibited non-enzymatic transacylation of glycine (85). Use of inhibitors like (-) borneol (inhibit acyl glucuronidation) and tri-methylacetic acid (TMA) (inhibit Acyl CoAs formation) in a mechanistic study of 2-Phenylpropionic acid (2-PPA) showed 49% reduction in covalent binding by inhibiting thioesters compared to just 23% of reduction by glucuronidation inhibition. This study shows thioesters are much more reactive than glucuronides and, thus, more important in covalent binding and subsequent toxicity (78). Furthermore, Naproxen-CoA and 2-PPA have shown 100-fold and 70-fold higher reactivity towards nucleophiles than their glucuronide antipodes respectively (78, 80). It has been demonstrated that reactivity of thioesters depends on the substitution at the α-carbon in the drug moiety. In the same study, a comparison of the reactivity of eight carboxylic acid drugs towards glutathione showed a linear co-relation between overall rate of hydrolysis and reactivity of acyl CoAs (86).
It could be argued that conjugation reaction to amino acid could be a step for cell self-protection as the reactive species formed endogenously can be inactivated by conjugating to amino acid by N-Acyl transferases. However, a problem occurs when reactive species acylate important proteins and form unusual drug-protein adducts, which cannot, readily, be excreted from the body or escape inactivation (76). Unfortunately, it is not yet possible to predict the target macromolecules. In addition, abnormal conjugation to proteins can alter vital functions such as signal transduction pathways and contribute to toxicity (55). Olsen and colleagues showed the production of Tolmetin CoA thioester in rat livers in vivo, which subsequently formed S-Acyl glutathione thioester conjugate (83). Valproic acid is shown to form liver protein adducts. However, as their acyl glucuronides are relatively stable and not very reactive towards proteins, it has been postulated that CoA thioesters may be involved in acylating proteins in this case (87).
In this context, modification of proteins should be considered to be an important underlying mechanism of IDRs. Studies show that acyl CoA thioesters are more reactive than acyl glucuronides. Thus, their involvement in conjugation reaction should be widely explored.
4.2 Stereo-selective inversion
The widely studied profen drugs are NSAIDs and are chiral carbon containing carboxylic acid moiety. These are mainly found as two R/S enantiomers (88). It has been well established that the anti-inflammatory action of profen drugs reside in (S)-enantiomer and the metabolic inversion of (R)-enantiomer to (S)-enantiomer acquired significance (88). Studies show that the formation of acyl CoA thioester is the first step in inversion of profens using ATP and CoA as co-factors (77, 88) (Fig. 7).
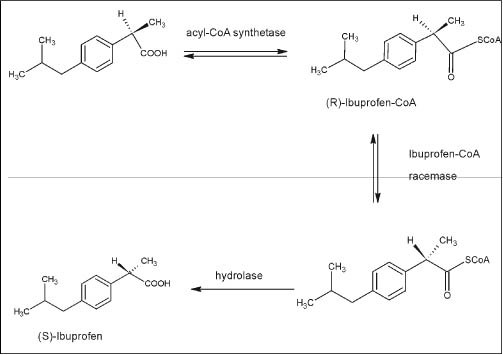
Figure 7. Metabolic stereo-selective inversion of (R)-Ibuprofen showing the formation of CoA derivative prior to the formation of (S)-Ibuprofen (89)
In addition to ibuprofen, several other profens have been studied for stereo-selective inversion in their chiral carbon in rats as well as humans (90-95). For each profen studied, the inversion was shown to be in favor of (R)-enantiomer rather than its antipodes. This strongly suggests that if there is a metabolic inversion of a carboxylic acid drug, acyl CoA thioester production is inevitable. The fact that Flurbiprofen, in a study, showed no production of thioesters when it underwent very little inversion, if any, supports this hypotheses (97). In another study, Flunoxaprofen demonstrates stereo-selective transacylation of proteins that is consistent with bioactivation by (R)-FLX-CoA formation (98). If acyl CoA thioesters are produced as an essential product of a metabolic pathway, their accumulation in the cells could result into toxic events, if they escape detoxification. Another aspect to be investigated is the production of additional quantities of (S)-enantiomers, which contribute to its therapeutic effects as well as the toxicity profile (79). However, the main fact highlighted by the stereo-selective inversion is that acyl CoA thioester formation is vital in the metabolic activation of most of the profen drugs. Furthermore, the production of acyl CoA thioesters can be co-related to the formation of drug-proteins adducts and associated toxicity, as aforementioned.
Stereo-selective inversion also shows species variability (98). For instance, the half-life of Benoxaprofen varies in animals and humans (99). Also, Ketoprofen readily shows inversion in rats but undergoes very little inversion in the human species (77). Chiral inversion is a general feature for the profen drugs. However, it shows a marked difference in its occurrence and rate for different species, which makes it difficult to extrapolate biological data from species to species and obstructs the development of a suitable animal model for predicting type B reactions.
4.3 Inhibition of specific cellular functions
In cellular metabolism, xenobiotic acyl CoA derivatives could interfere with the normal physiological activity of the cell by competing with endogenous acyl CoA’s functions and by acting as substrates for acyl CoA ligase, as mentioned earlier in the case of ibuprofen and palmitic acid (77). On the outer membrane of mitochondria, fatty acids are converted to acyl CoAs. These convert further to acyl-carnitine esters by carnitine-acyl transferase (CAT I), which allows for the transport of fatty acids across the mitochondrial membrane. On the inner mitochondrial membrane, acyl-carnitine esters are converted into acyl CoA thioesters by CAT II, subsequently oxidizing fatty acids and generating acetyl CoA and ATP (100). Formation of xenobiotic acyl CoAs may disrupt this pathway by sequestering either CoA or carnitine or by inhibiting the enzyme involved in β-oxidation (101). Furthermore, the level of CoA in cells is relatively low and utilisation of CoA by exogenous carboxylic acid limits the availability of CoA for other pathways such as pyruvate oxidation, krebs cycle, lipid synthesis and post translational modification by fatty acids (102-103). Salicyl CoA thioester showed, in vitro, depletion of mitochondrial CoA, as increasing the concentration of CoA in incubations ameliorated the inhibition of β-oxidation of palmitic acid otherwise observed (104). Diclofenac is known to interrupt normal mitochondrial function, minimise ATP production and causes mitochondrial swelling (105, 69). It generates oxygen reactive species and is involved in oxidation of the protein thiol groups (69). Noticeably, Diclofenac is more cytotoxic to drug metabolising cells than non-metabolising cells (79). With NSAIDs, hypolidaemic drugs also show a similar deterioration of enzymatic pathways related to the production of thioesters (106). Endogenous acyl CoAs are involved in regulating key enzymes like citrate synthetase (107) and glucokinase (108). The intrusion of xenobiotic acyl CoAs might disturb this regulation.
Valproic acid causes microvesicular steatosis and necrosis and is presumed to be toxic due to the accumulation of its thioesters in mitochondria, which are impermeable to the inner mitochondrial membrane and cannot easily be excreted from cells. (109-111). Similarly, it has been reported that the accumulation of L-2-amino-3-methylenecyclopropylpropionic acid (MCPAA) CoA conjugate in cells has resulted in cases of human poisoning which enhances the reputation of CoA thioesters as being toxic metabolites (102).
4.4 Incorporation in glycerolipids
The role of CoA thioesters in forming unusual hybrid lipids by being incorporated into endogenous lipids like diglycerols, triglycerols and phospholipids is well established (112-116). The literature shows that there is also evidence of the incorporation of NSAIDs like ibuprofen (116) and fenoprofen (117). Hypolipidaemic agents like 4-BenzyloxyBenzoic acid (BRL 14280) (113) being formed into lipid molecules has been shown in rats and humans. The consequences are not clearly understood but it was noted that the production of hybrid lipids is associated with the inhibition of lipid synthesis (118). A comparative study shows that fenoprofen has better capacity to form hybrid lipids and inhibition of lipid synthesis in rats than ibuprofen and flurbiprofen (115). One of the other plausible toxic effects of hybrid lipids is the perturbation of protein kinase C activity and cell signal transduction by xenobiotic containing di-acylglycerols (77). Hybrid lipid formation lengthens the period that drugs remain inside the body making it like a reservoir for further pharmacological action (115). It could, also, enter into the cell, alter the microenvironment for the regulating enzymes, and impair cell viability (113). (R)-enantiomers of ibuprofen have been shown to be more effectively incorporated into the lipids than their antipodes (114), suggesting the significance of stereo-selectivity in forming hybrid lipids (119). Not much information about their formation in humans is available but a study showing the metabolism of pivalic acid (a component of Pivaloyloxyethyl, a prodrug of methyldopa) via the acyl CoA formation pathway and not by glucuronidation, as seen in rats, monkeys and rabbits, suggests that humans might be particularly susceptible (120).
5.0 Discussion
The above discussion shows that carboxylic acid-derived metabolites could lead to adverse events in body. Careful handling of such drugs during the discovery phase and the need of effective experimental approaches to detect the production of reactive metabolites is essential. Current animal models cannot predict the production of acyl thioesters in new candidate drugs; therefore, the development of an assay system to be used pre-clinically, to enhance the risk assessment of these drugs prior to entry into clinical trials, is required. A possible approach to the design of such an assay system would be to develop a strategy that could demonstrate, in vitro, whether protein conjugation, mainly with glycine, of a substrate is possible (provided all the necessary cofactors and reactants are present) and to what degree, in each test system. Amino acid conjugation is a two-staged reaction in which the first step is the formation of acyl CoA thioester by acyl CoA synthetase followed by transfer of the acyl group to glycine by N-acyl transferases (121). The investigation of protein conjugates would then act as a biomarker to propose the production of thioesters.
There are a number of drugs and acids that have been found as substrates for CoASH, but limited work has been done to explore the conjugation reaction with amino acids in vitro/in vivo (103). However, if each reaction is considered separately, it would appear that amino acid conjugation helps in ‘moping out’ highly reactive intermediates to maintain the mitochondrial integrity. Therefore, it would be logical to assume that xenobiotic thioesters, which are not the substrate for endogenous amino acid conjugation, would be a mitochondrial toxin. It is apparent from the discussion so far that acyl CoA thioesters are reactive intermediates which could be inactivated by amino acid conjugation. They could lead to sequestration of CoASH in the mitochondrial matrix and cause potential harm to biological cycles; hence toxicity. The other enzyme to consider in amino acid conjugation is N-acyl transferases as it carries out the second step of transferring the acyl group to glycine and releases CoA. Both the enzymes have been specifically recognised for chemical moieties and are responsible for the comprehensive study of the metabolism of xenobiotic acid via amino acid conjugation. Although a limited number of structure–metabolism studies have alluded to the subtle nature of the molecular recognition of the medium-chain CoA ligase, these studies have provided no insight into the factors influencing the recognition of the xenobiotic acyl-CoA by the glycine N-acyltransferases (103). Obviously, further elucidation of the critical structural aspects of substrates for both the medium-chain CoA ligase and the N-acyltransferase is essential to enable pharmaceutical companies to make prudent decisions regarding the likelihood of mitochondrial toxicity from new chemical entities. Clearly, as studies have demonstrated, acyl CoA thioesters are more reactive than glucuronides but glucuronides are formed in higher concentrations. Unlike glucuronides, thioesters cannot be excreted from the cells and can accumulate in mitochondria as exemplified by the formation of thioesters of tolmetin, hypoglycine and valproic acid (122-125). Mitochondrial functions are vital to the cells and their dysfunction could be more detrimental than a non-specific modification of proteins.
Another thing to consider is that IDRs occur in certain subtypes and not in every individual; detecting reactive metabolites could give an idea of their occurrence. However, problems persist as the data is generated from a limited source and there cannot be certainty whether production of reactive electrophilic species would always show idiosyncratic reactions. To combat this, the use of induced pluripotent cells from the affected individuals to deduce the metabolic fate of compounds could also be seen as a potential option in future. It would give an idea of the differences and other factors involved in type-B drug reactions, as these reactions are highly individual specific. It would be helpful to obtain robust data specific to such reactions.
In conclusion, based on the aforementioned evidence, it is time that equal attention is paid to acyl CoA thioesters as reactive, potentially toxic, metabolites and they should be handled as ‘structural alerts’ in causing adverse drug reactions.
References
Kola, I. and Landis, J. (2004) Nature Reviews Drug Discovery, 3,711-715.
DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G and L. Lasagna, L. (1991) J. Health Econ., 10, 107–142.
DiMasi, J.A.; Hansen, R.W. and Grabowski, H.G. (2003) J. Health Econ., 22, 151–185.
Adams, C.P. and Brantner, V.V. (2010) J. Health Econ., 19, 130–141.
Hughes, B. (2010) Nature Reviews Drug Discovery, 9, 89-92 .
Kalgutkar, A.S. and Didiuk, MT (2009) Chemistry and Biodiversity, 6, 2115-2137.
[No author listed] (2011) Nature Rev. Drug Discovery, 10, 7-9.
Borzelleca, J.F. (2001) in principles and methods of toxicology 4th Ed. (Hayes, A.W. Ed.) Taylor and Francis, Philadelphia, pp. 1-21.
Borzelleca, J.F. (2000) Toxicol.Sci., 53, 2-4.
W.H.O. (1972) Tech. Rep. Ser. WHO, no. 498.
Miller, R.R. (1974) Arch. Intern. Med., 134, 219–223.
Wiffen P.; Gill M.; Edwards J. and Moore A. (2002) Bandolier Extra, June, 1-16.
Smith, C.C.; Bennett, P.M.; Pearce, H.M.; Harrison, P.I; Reynolds, D.J.; Aronson, J.K. and Grahame-Smith, D.G. (1996) Br. J. Clin. Pharmacol., 42, 423-9.
Auzepy, Ph.; Durocher, A.; Gay R., et al (1979) Nouv. Presse. Med., 8, 1315-8.
Asscher, A.W.; Parr, G.D. and Whitmarsh, V.B. (1995) British Medical Journal, 311(7011), 1003-1006.
Lazarou, J.; Pomeranz, B.H. and Corey ,P.N. (1998) Journal of the American Medical Association, 279, 1200-1205.
Wester, K.; Jonsson, A.K.; Spigset, O.; Druid, H. and Hagg, S. (2008) Br. J. Clin. Pharmacol., 65(4), 573-579.
Bakka, O.M.; Manocchia, M.; de Abajo, F., et al. (1995) J. Clin. Pharmacol. Ther., 58, 108-117.
Holt, M.P. and Ju, C. (2006) American Association of Pharmaceutical Scientists Journal, 8, E48-E54.
Park, B.K.; Pirmohamed, M. and Kitteringham, N.R. (1992) Br. J. clin. Pharmac., 34, 377-395.
Bates, D.W.; Spell, N.; David, J.; Cullen, D.J.; Burdick, E.; Laird, N.; Petersen, L.A., et al (1997) Journal of the American Medical Association, 277(4), 307-311.
Zimmerman, H.J. (1978) Drugs., 16(1), 25-45.
Timbrell, J.A. (1983) Br. J. clin. Pharmac., 15, 3-14.
Park, B.K.; Kitteringham, N.R.; Maggs, J.L.; Pirmohamed, M. and Williams D.P. (2005) Annual Review of Pharmacology and Toxicology, 45, 177-202.
Park, B.K.; Kitteringham, N.R.; Powell, H. and Pirmohamed M. (2000) Journal of Toxicology, 153, 39–60.
Lasser, K.E.; Allen, P.D.; Woolhandler, S.J.; Himmelstein, D.U.; Wolfe, S.M. and Bor, D.H. (2002) Journal of the American Medical Association, 287, 2215-2220.
Caldwell, J.; Hutt, A.J. and Fournal-Gigleux, S. (1988) Biochem. Pharmacol., 37, 105-114.
Baillie, T.A. (1992) Pharm. Weekbl. Sci., 14, 122-125.
Lygre, T.; Aarsaether, N.; Stensland, E.; Aarsland, A. and Berge R.K. (1986) J. Chromatogr., 381, 95-105.
Sabordo, L.; Sallustio, B.C.; Evans, A.M. and Nation R.L. (2000) J. Pharmacol. Exp. Ther., 295, 44-50
Wilman, D. Duract: Painkiller posed risk of damage to liver. The LA Times. Dec 20, 2000.
Barozzi, N. and Tett, S.E. (2009) Clin. Rheumatol., 28, 509–519.
Pierson, R.N. Jr.; Holt, P.R.; Watson, R.M. and Keating, R.P. (1993) Br.J.Clin.Pharmac., 38, 219-226.
Bjorkman, D. (1998) The American Journal Of Medicine, 105(5), 17S-21S.
Lewis, J.H. (1984) J. Clin. Pharm., 3(2), 128-38.
Rabinovitz, M. and VanThiel, D.H. (1992) Am. J. Gastroenterol., 87, 1696-1704.
Boelsterli, U.A. (2002) in Drug-Induced Liver Disease, (N. Kaplowitz and L. DeLeve Eds.), Marcel Dekker, New York, NY, pp. 345-375.
Carson, J.L.; Storm, B.L.; Duff, A., Gupta, A and Das, K. (1993) Arch. Interm. Med., 153, 1331-1336.
Murray, M.D. and Brater. D.C. (1993) Annu. Rev. Pharmacol. Toxicol., 33,434-465.
Zimmerman, H.J. (1994) in Nonsteroidal Anti-inflammatory Drugs, (A.J. Lewis and D.E Furst Eds.), Marcel Dekker, New York, pp. 171-194.
Gracia Rodriguez, L.A.; Williams, R.; Derby, L.E.; Dean, A.D. and Jick, H. (1994) Arch. Intern. Med., 154,311-316.
Singh, G.; Ramey, D.R.; Morfeld, D. and Fries, J.F. (1994) Pharmacol. Ther., 62, 175-191.
Boelsterli, U.A.; Zimmerman, H.J. and Kretz-Rommel, A. (1995) Crit. Rev. Toxicol., 25(3):207-235.
Palmer, W.L. and Woodall, P.S. (1936) Journal of the American Medical Association, 107,760.
Lewis,J.H. (1998) Clinics in liver disease, 2(3), 543-561.
Bartle, W.R.; Gupta, A.K. and Lazor, J. (1986) Arch Intern Med., 146(12), 2365-2367.
Evans, D.C.; Watt, A.P.; Nicoll-Griffith, D.A. and Baillie, T.A. (2004) Chem Res Toxicol., 17, 3-16.
Pirmohamed, M. and Park k. (2001) TRENDS in pharmacological sciences, 22(6), 298-305.
Woodson, L.C.; Dunnette, J.H. and Weinshilboum, R.M. ( 1982 ) J. Pharmacol. Exp. Ther., 222 ( 1 ), 174 – 181.
Liebler, D.C. and Guengerich, F.P (2005) natures review., 4, 410-420.
Park, B.K.; Kitteringham, N.R. and Pirmohamed M. (1992) Br.J. clin. Pharmac., 34, 377-395.
Ponsoda, X.; Pareja, E.; Gómez-Lechón, M.J.; Fabra, R.; Carrasco, E.; Trullenque, R. and Castell, J.V. (2001) J. Hepatol., 34(1), 19-25.
Lee W.M. (1995) the New England Journal Of Medicine, 333 (17), 1118-1127.
Evans, D,C,; Watt, A.P.; Nicoll-Griffith, D.A. and Baillie, T.A. (2004) Chem. Res. Toxicol., 17, 3-16.
Boelsterli, U.A. (2002) Current drug metabolism, 3, 439-450.
Fura A,; Shu YZ,; Zhu M,; Hanson RL,; Roongta V. and Humphreys WG (2004) J. Med. Chem., 47, 4339-51.
Hutt, A.J. and Caldwell, J. (1990) In Amino acid conjugation 1st Ed. (Mulder Gj, Ed.) Taylor and Francis, London, U.K. p.p 273-304.
Dixon, P.A.F.; Caldwell, J. and Smith, R.L. (1977) Xenobiotica, 7, 695-706
Spahn-Langguth, H. and, Benet L.Z. (1992) Drug Metab. Rev., 24, 5-47.
Boelsterli, U.A. (2002) in Mechanisms, Models and Predictions of Idiosyncratic Drug Toxicity, (V. Subrahmanyan Ed.), ISE Press, Brentwood, MO, in press.
Skonberg, C.; Olsen, J.; Madsen, K.G.; Hansen, S.H. and Grillo M.P. (2008) Expert Opin. Drug Metab. Toxicol., 4(4), 425-438.
Boelsterli, Urs.; Ramirez-Alcantara and Veronica (2011) Current Drug Metabolism (12) 245-252
Burchell, B.; McGurk, K.; Brierley, C.H. and Clarke, D.J. (1997) In comprehensive Toxicology, (I.G. Sipes, C.A. McQueen and A.J. Gandolfi Eds.) Elsevier, New York, pp.401-435.
Soars, M.G.; Railey, R.J.; Findlay, K.A.B.; Coffey, M.J. and Burchell, B. (2001) Drug Metab. Dispos., 29, 121-126.
Seitz, S. and Boelsterli, U.A. (1998) Gastroenterology, 115, 1476-1482.
Bort, R.; Ponsoda, X.; Jover, R.; et al (1999) J. Pharmacol. Exp. Ther., 288, 65–72.
Kretz- Rommel, A.; Boelsterli, U.A. (1995) J. Hepatology, 22, 213-22.
Masubuchi, Y.; Nakayama, S. and Horie, T. (2002) J. Hepatology., 35, 544–51.
O’connor N.; Dargan, P.I. and Jones A.L. (2003) QJM: An International Journal of Medicine, 96(11), 787-791.
Wang, M.; Gorrell, M.D.; Mc Gaughan, G.W., et al (2001) J. Life Sci., 68, 785-97.
Wang, M.; Gorrell, M.D.; Abbott C.A., et al (2002) J. Gastrointerol Hepatol., 17, 66-71.
Dong, J.q.; Liu, J. and Smith, P.C. (2005) Biochem. Pharmacol., 70, 937-48.
Killenberg, P.G.; Davidson, E.D and Webster, L.T., Jr. (1971) Mol. Pharmacol., 7, 260-268
Knights, K.M. (1998) Clin. Exp. Pharm. Physiol., 25, 776-782
Londesborough, J.C. and Webster, L.T. (1974) In Fatty acyl-CoA synthetases, (Boyer, P.D. Ed.) New York: Academic Press, p.p. 469-488.
Knights, K.M. and Drogemuler, C.J. (2000) Current Drug Metab., 1, 49-66.
Hall, S.D. and Xiaotao, Q. (1994) Chem. Biol. Interact., 90, 235-251.
Tracey, T.S.; Wirthwein, D.P and Hall, S.D. (1993) Drug Metab. Dispos., 21, 114-120.
Li, C.; Grillo, M.P. and Benet, L.Z. (2002) J. Pharmacol. Exp. Ther. 305(1), 250-6.
Olsen, J.; Li, C.; Bjornsdottir, I.; Sidenius, U.; Hansen, S.H. and Benet, L.Z. (2005) Chem. Res. Toxicol., 18 (11), 1729–1736.
Olsen, J.; Bjornsdottir, I.; Tjornelund, J. and Hansen, S.H. (2002) J. Pharm. Biomed. Anlysis, 29, 7–15.
Lygre, T.; Aarsaether, N.; Stensland, E.; Aarsland, A. and Berge, R.K. (1986) J. Chromatogr., 381, 95-105.
Olsen, J.; Li, C.; Skonberg, C.; Bjornsdottir, I.; Sidenius, U.; Hansen, S.H. and Benet, L.Z. (2007) Drug Metab. Dispos., 35, 758-764.
J, potter. (1957) Physiol.Chem., 308, 81-90.
Tishler, S.L. and Goldman, P. (1970) Biochem. Pharmacol., 19, 143-50.
Sidenius, U.; Skonberg, C.; Olsen J. and Hansen, S.H. (2004) Chem. Res. Toxicol., 17, 75-81.
Bailey, M.J and Dickinson, R.G (1996) Chem. Res. Toxicol., 9, 659-666.
Li, C.; Benet, L.Z. and Grillo, M.P. (2002) Chem. Res. Toxicol., 15 (10), 1309-1317
Carvalho, P.O.; Q. B. Cass, Q.B.; S. A. Calafatti, S.A.; F. J. Contesini, F.J. and Bizaco, R. (2006) Braz. J. Chem. Eng., 23(3), 291 – 300.
Boop, R.J.; Nash, J.F.; Ridolfo, A.S. and Shepard, E.R. (1979) Drug Metab. Dispos., 7, 356-9.
Rubin, A.; Knadler, M.P.; Ho, P.P.K.; Bechtol, L.D. and Wolen, R.L. (1985) J. Pharm.Sci., 74, 82-84
Abas, A. And Meffin, P.J. (1987) J. Pharmacol. Exp. Ther., 240, 637-641.
Goto, J.; Goto, N. And Nambara, T. (1982) J. Chromatog., 239, 559-564.
Tamassia, V.; Jannuzzo. M.G.; Moro, E.; Stegnjaich, S.; Groppi, W. and Nicolis, F.B. (1984) Int. J. Clin. Pharm. Res., 4, 223-230.
Fournal, S. And Caldwell, J. (1986) Biochem. Pharmacol., 35, 4153-4159.
Knadler, M.P. and Hall, S.D. (1990) Chirality., 2(2), 67-73.
Grillo, M.P.; Wait, J.C.; Lohr, M.T.; Khera, S. and Benet, L.Z. (2010) Drug Metab. Dispos., 38, 133-142.
Caldwell, J.; Hull, A.J Fournel-Gigleux, S. (1988) J. Biochem. Pharmacol., 37(1), 105-114.
Hutt, A.J. and Caldwell, J. (1983) Journal of Pharmacy and Pharmacology, 35(11), 693-704.
Biochemistry, 2nd Ed., Eds. Mathews, C.K. and Van Holde, K.E., The Benjamin/Cummings Publishing company, InC., Menlo Park, CA, 1995, ISBN 0-8053-3931-0
Fromentry, B. and Pessayre, D. (1995) Pharmacology & Therapeutics, 67 (1), 101-154.
Brass, E.P. (1994) Chem. Biol. Interact., 90, 203-214.
Knight,K.M.; Sykes, M.J. and Miners, J. (2007) Expert opin. Drug Metab. Toxicol., 3(2), 159-168.
Deschamps, D.; Fisch, C.; Fromenty, B.; Berson, A.; Degott, C. and Pessayre, D. (1991) J. Pharmacol. Exp. Ther., 259 (2), 894-904.
Boesterli, U.A. (2003) Toxicol. Appl. Pharmacol., 192, 307-22.
Miguel Bronfman, M.; Amigo, L. and Morales, N.M. (1986) Biochem. J., 239, 781-784.
Hansel, B. C. & Powell, G. L. (1984) J. Biol. Chem., 259,1423-1430.
Tippet, P. S. & Neet, X. E. (1982) J. Biol. Chem., 257,12839-12845.
Silva, M.F.; Ijlst, L.; Allers, P.; Jakobs, C.; Duran, M.; de Almeida, I.T.and Wanders, R.J. (2004) Drug Metab. Dispos., 32 (11) 1304-1310
Bjorge, S.M. and Baillie, T.A. (1985) Biochem. Biophys. Res. Commun., 132 (1), 245-252.
Silva, M.F.B.; Ruiter, J.P.N.; Ijlst, L.; Allers, P.; ten Brink, H.J.; Jakobs, C.; Duran, M.; de Almeida, I.T. and Wander, R.J.A (2001) Analytical Biochemistry, 290 (1) 60-67.
Dodds, P.F. (1991) Life Sci., 49(9), 629-49.
Fears, R.; Baggaley, K.H.; Alexander, R.; Morgan, B. And Hindley, R.M. (1978) J. Lipid Res., 19, 3-11.
Moorhouse, K.G.; Dodds, P.F. and Hutson, D.H. (1991) Biochem. Pharmacol., 41(8), 1179-1185.
Fears, R. (1985) Prog. Lipid Res., 24, 177-195.
Williams, K.; Day, R.; Knihinicki, R and Duffield, A. (1986) biochem. Pharmacol., 35, 3403-3405.
Sallustio, B.C.; Meffin, P.J. and Knights, K.M (1988) Biochem. Pharmacol., 37, 1919-1923.
Fears, R. and Richards, D. H. (1981) Biochem. Sot. Trans., 9, 572-573.
Haybell, P.J and Meffin, P.J. (1987) J. Phamracol. Exp. Ther., 240, 631-636.
Vickers, D.; White, C.; Ramjith, S.D.; Smith, G.; Walker, J.L.; Flynn, R.W. and Arison. B. H. (1985) Xenobiotica, 15, 453-458.
Mahler, H.R.; Wakil, S.J. and Bock, R.M. (1953) J.Biol. Chem., 204, 453-468.
Tatsuhara, T.; Muro, H.; Matsuda, Y.; et al. (1987) J. Chromatogr., 399, 183 -95.
Wenz, A.; Thorpe, C. and Ghisla S. (1981) J. Biol. Chem., 256, 9809 -12.
Lieu, Y.K.; Hsu, B.Y.; Price, W.A.; et al. (1997) J. Physiol. 272, E359 -66.
Olsen J. Reactive drug metabolites: formation, metabolism and reactivity of acyl coenzyme A thioesters In Vitro and in vivo. PhD thesis, Department of Analytical and Pharmaceutical Chemistry, Royal Danish School of Pharmacy, Copenhagen; 2002