The Role of Glutamate Receptors in Excitotoxicity during Stroke
Introduction:
- Glutamate receptors mediated excitotoxicity is a major consequence of stroke, which ultimately leads to neuronal death resulting in cerebral damage (Hazell, 2007). Stroke (also known as brain attack or cerebrovascular accident) is a life-threatening event, in which part(s) of brain is deprived of adequate oxygenated blood and glucose. Stroke is the 2nd (or 3rd) leading cause of death in industrialised countries (Moro, et al., 2005), especially in western countries. In 1999, about 5.5 million people died worldwide from stroke, which is approximately 10% of all deaths (Lo, et al., 2003). Stroke initiates neuronal death resulting from over-excitation of glutamate receptors, which leads to excessive release of glutamate (Ikeda, et al., 1996). Excessive glutamate release triggers an overload of intracellular Ca2+ through NMDA receptors and voltage-gated Ca2+ channels ((MacDonald, et al., 2006). This excessive intracellular Ca2+ leads to excitotoxicity and subsequent cell death in the brain.
- Stroke:
- Stroke, as mentioned earlier, is the leading cause of death and disability worldwide. Stroke can be ischemic (due to interruption of blood supply, in the form of clots) or hemorrhagic (due to rupture of blood vessels / abnormal vascular structure / bleeding in or around the brain). Ischemic strokes are more common, accounting for 80% of the total number of strokes. Ischemic strokes are really life-threatening. The brain does not get enough blood (and so enough oxygen and glucose) in such a condition, due to a blockade of the blood supply (generally due to the clot formations). This leads to hypoxic condition, resulting in excessive glutamate release. The clots in the cerebral artery may be caused by ‘hardenings of the arteries’ in the carotid artery that feeds the head and brain with oxygen rich blood. (http://yourtotalhealth.ivillage.com/stroke?sky=ggl|ths|plavix|stroke|)
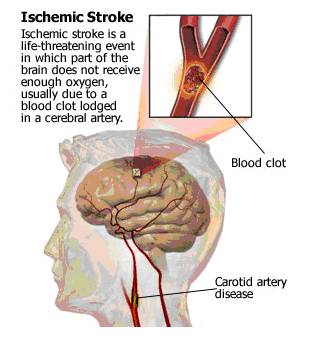
- Fig. 1: Figure depicting the development of ischemic stroke due to clot formation. [Taken from http://yourtotalhealth.ivillage.com/stroke-basics.html?pageNum=2#2]
- Excitotoxicity:
- Following ischemic stroke excitotoxicity leads to neuronal cell death. Excitotoxicity is a key mechanism of cell and tissue destruction in cerebral ischemia. The phenomenon of excitotoxicity was discovered by D R Lucas and J P Newhouse in 1957 (Purves, et al., 2008. pp 129). Excitotoxicity refers to the pathological process by which over-activations of the excitatory neurotransmitter (glutamate and related compounds like aspartate) lead to neuronal damage. Glutamate is the major excitatory neurotransmitter in the mammalian CNS. Glutamate mediates its action by binding to both ionotropic and metabotropic receptors. The glutamate receptors that are located at post synaptic terminals, mediate depolarisation and Ca2+ entry into the cells to trigger excitotoxicity, on excessive exposure to glutamate.
- Glutamate receptors are mainly of two kinds, viz. ionotropic and metabotropic. The ionotropic receptors are further classified into three types- NMDA, AMPA and kainite receptors.
- Ionotropic Glutamate Receptors:
- The AMPA and Kainate receptors are highly permeable to Na+ and K+ ions, but little or not permeable to the Ca2+ ions. AMPA receptors allow Ca2+ ions to pass only if the receptors lack the GluR2 subunit (Modi and MacDonald, 1995). AMPA receptors are responsible for fast synaptic transmission and the initial component of the EPSP. AMPA receptors require only glutamate to get activated. On the contrary NMDA receptors are highly permeable to Ca2+, Na+ ions and K+ ions as well. The NMDA receptors are responsible for slow synaptic transmission and the late phase of the EPSP. The NMDA receptors require both glutamate and depolarisation in order to get activated. During resting membrane potential the NMDA-channel is blocked by Mg2+ ion, and to be relieved from that blockade NMDA receptors require depolarisation, which is provided by the AMPA receptors. Since the AMPA receptors are permeable to the monovalent cations, Na+ ions freely enter the cells along its electrochemical gradient and triggers depolarisation. This depolarisation (in the post synaptic membrane) relieves Mg2+ block and the opens the voltage-gated calcium channels as well. (Dirnagl, et al., 1999; Lo, et al., 2003; Hazell, 2007; http://www.bris.ac.uk/Depts/Synaptic/info/glutamate.html)
Metabotropic Glutamate Receptors:
Metabotropic glutamate receptors (mGluRs) are G-protein coupled receptors, which produce their effects via phosphoinositides, cAMP and other second messengers. mGluRs located at the post-synaptic nerve terminals (Group I mGluRs) produce slow depolarisation by releasing Ca2+ from the intracellular stores; while those located at the pre-synaptic nerve terminals (Group ii mGluRs) inhibit glutamate release by inhibiting adenylyl cyclase (Hazell, 2007).
Roles of Glutamate Receptors in Excitotoxicity following a Stroke:
Now, there are many processes in which glutamate receptors trigger excitotoxicity during ischemic stroke, and those are overlapping as well, some occurring simultaneously. Brain tissue requires a huge supply of oxygen and glucose due to the voracious metabolic rate, and the energy supply depends on oxidative phosphorylation. Neuronal activity stops on cessation of blood flow due to termination of supply of oxygen and glucose.
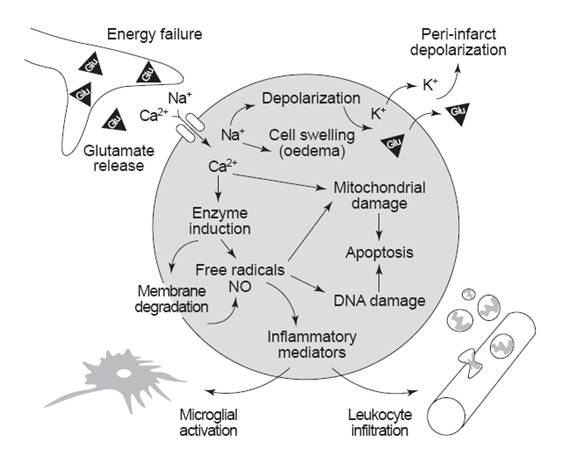
Fig. 2: Simplified overview of the roles of glutamate receptors in causing excitotoxicity following ischemic stroke. [Dirnagl, et al. (1999). Trends in Neurosciences. 22(9): 393]
During stroke the blood supply and thus the substrates (oxygen and glucose) supply is impaired. At this point, neurons cannot generate enough ATP to keep their ion pumps working properly. Due to this energy depletion, the resting membrane potential gets interfered, which leads to depolarisation of neurons and glia, and subsequent release of Ca2+ into cells (Bear, et al., 2007. pp 156). Increase in intracellular Ca2+ triggers synaptic release of glutamate. Excess glutamate further depolarise cells and thereby raises the intracellular Ca2+ concentration. Consequently further release of glutamate occurs. Thus, stroke initiates a vicious cycle of excess glutamate release. At this point, excessive glutamate in the extracellular space leads to the following extensively interactive and overlapping excitotoxic events:
Interference with the Glutamate Transport:
Downregulation of glutamate transporters: Due to cerebral ischemia downregulation of glutamate transporter genes- EAAT1 and EAAT2 occurs (Hazell, 2007). Thus increased glutamate release takes place.
Reversal of glutamate efflux: Due to energy depletion and uncontrolled release of glutamate, the ATP levels decreases in the astrocytes, resulting in the failure of the Na+-K+-ATPase pump, which leads to the collapse of the ionic gradient. As a result, the Na+ ions move along its concentration gradient into the cell, while glutamate is released into the extracellular space. This reverse uptake further enhances the accumulation of extracellular glutamate, thus facilitating the development of excitotoxicity. (Moro, et al., 2005; Hazell, 2007)
Vesicular release of glutamate: Though astrocytes are meant for removal of excess glutamate from the extracellular space, during the ischemic stroke due to increased intracellular Ca2+ levels, the astrocytes are enabled to release glutamate in the extracellular space via a vesicular-mediated process.
(Hazell, 2007)
Ionic Imbalance:
Excessive accumulation of extracellular glutamate leads to overactivation of glutamate receptors (NMDA) and results into entry of high amounts of Na+ and Cl- ions through the receptor-channels (AMPA). Along with these ions water also enters the cells due to an increase in osmolality. This total influx is much larger than the efflux of K+ ions. This results in excessive swelling and neuronal oedema. This extreme swelling finally leads to lysis of the neurons. This is termed as ‘initial toxicity’, which involvesincreased sodium permeability that results in depolarisation, chloride entry and eventually, osmotic lysis of swollen cells. This is followed by delayed toxicity due to Ca2+ entry (through glutamate receptor-channels and/or voltage-gated Ca2+ channels). (Rothman and Olney, 1986; Choi, 1987 Dirnagl, et al., 1999; Lo, et al., 2003; Hazell, 2007).
Generation of Reactive Oxygen Species (ROS):
During ischemic stroke, generation of ROS is a significant event, which then leads to damage of lipids, proteins, DNA and consequent neuronal damage (Moro et al., 2005). Due to energy depletion glutamate is released at high concentrations ‘within the core of cerebral infarction and at the penumbral tissue’ (Dirnagl, et al., 1999). In these areas, over-activation of glutamate receptors leads to a massive calcium influx. This results in activation of several calcium-dependent enzymes like calpain, proteases, phospholipases, endonucleases etc (Moro, et al., 2005). This is followed by a generation of NO, free radicals and reactive oxygen species (ROS) (like super oxide radical anion, hydroxyl free radical and powerful oxidant peroxynitrite) (Hazell, 2007). They promote damage of lipids (lipolysis), mitochondria and DNA. All these lead to neuronal death through apoptosis. ROS plays a significant role in the process of delayed neuronal death through various cellular signalling pathways, among which mitogen-activated protein kinase (MAPK) pathway (which involve persistent activation of extracellular signal-regulated kinases -ERKs) is a significant one (Stanciu, et al., 2000).
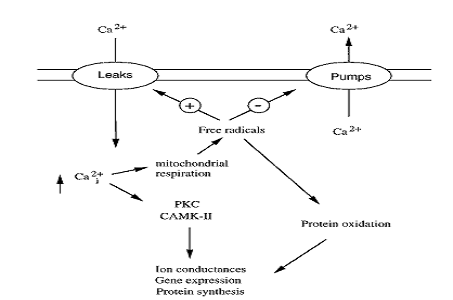
Fig. 3: Figure showing the coupling between calcium-induced signal transduction and calcium-triggered production of free radicals. [Kristian and Siesjo (1998). Stroke. 29: 712]
Oxidative Stress and Mitochondrial & DNA Damage:
As discussed earlier, following stroke massive glutamate release results into very strong activation of glutamate receptors (particularly the NMDA receptors). This results in an increase in ‘intracellular calcium levels, free radicals production, transient depletion of extracellular calcium through NMDA receptors and voltage-gated calcium channels’ (MacDonald, et al., 2006). There are two phases of calcium influx, and the second one, being very slow, is responsible for delayed neuronal damage (Beilharz, et al, 1995). The initial phase is the cause and effect of overactivation of NMDA receptors, which lead to osmotic swelling and subsequent cell lysis. The second phase is mainly responsible for mitochondrial calcium overload and damage of nucleic acids. Calcium, as discussed earlier, generates NO and ROS through activating enzymes like phospholipases, endonucleases, and proteases. The free radicals/ROS are generated either by the PLA2 activity or through the calcium-dependent NOS pathway. Mainly the second route is responsible during ischemic stroke. (Kristian and Siesjo, 1997; Stanciu, et al., 2000; Lo, et al., 2003)
Glutamate-mediated Apoptosis:
During ischemia excessive glutamate is also responsible for interfering with the activity of anti-apoptotic gene (Bcl-2) (Purves, et al., 2008. pp 646-647). Inactivation or suppression of the Bcl-2 allows cytochrome-c to be released by the mitochondria (Purves, et al., 2008. pp 646-647). This leads to the activation of caspase-3 (via caspase-9 activation) thus resulting in apoptosis and phagocytosis of the downstream molecules. (Ikeda, et al., 1996; Lo, et al., 2003)
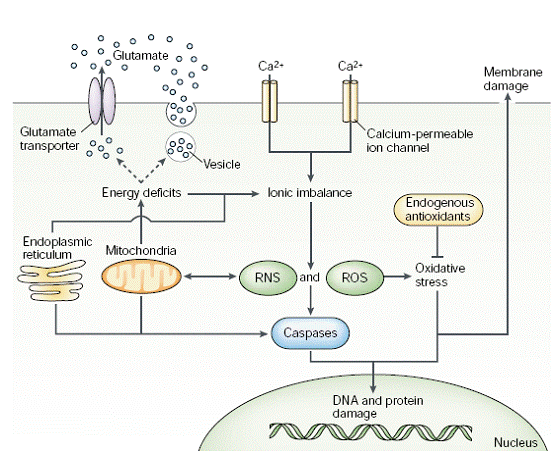
Fig. 4: Figure illustrating the interaction and overlapping between multiple pathways of excitotoxicity that lead to cell death in the brain. [Lo, et al. (2003). Nature Reviews Neuroscience. 4: 400]
Peri-infarct Depolarisations:
Following an ischemic stroke, glutamate as well as potassium ions are in high concentration in the extracellular fluid. In the core region and surrounding regions of ischemia affected areas, repetitive spreading depression like depolarisations occur, triggered by anoxic release of glutamate and K+ ions. They are in a situation in which they can depolarise continuously but cannot repolarise. These repetitive depolarisations are called peri-infarct depolarisations. The size of the infarct increases with the increase in frequency or number of depolarisations. These repetitive depolarisations are very much significant in facilitating glutamate mediated excitotoxicity. (Dirnagl, et al., 1999)
Upregulation and Downregulation of Certain Genes:
Following an ischemic stroke, upregulation or downregulation of specific genes contribute in some ways to the excitotoxicity and cell damage.
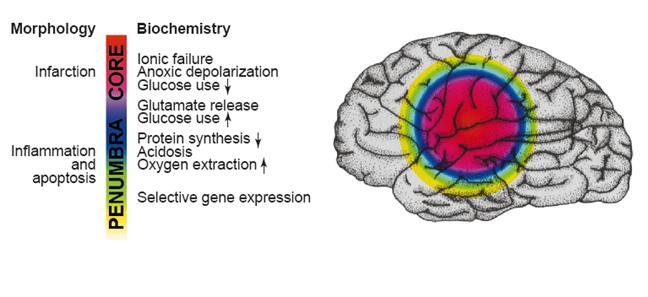
Fig. 5: Figure explaining the ischemic penumbra and the biochemical events in different layers in the brain. [Dirnagl, et al. (1999). Trends in Neurosciences. 22(9): 394]
For instance, after stroke there is a reduction in the Ca2+-impermeable GluR2 subunits of the AMPA receptor (Lo, et al., 2003). In some cases, downregulation of the Group I and II metabotropic glutamate receptors have also been reported. The exact causes are not known for these gene regulations, but all these favour the release of excessive glutamate. Apart from those genes, some inflammation-related genes are also upregulated which further increases in cell injury (Dirnagl, et al., 1999).
White Matter Injury:
Excitotoxicity in the white matter is slightly different from that in the grey matter in the brain (Lo, et al., 2003). The main components of the white matter are oligodendrocyte, endothelial cell, astrocyte and myelin. In the white matter there is synapse, so no vesicular release of glutamate occurs. Instead of that reverse glutamate efflux occurs in axons and oligodendrocytes (Lo, et al., 2003). Oligodendrocytes are damaged by excess glutamate, whereas axons are damaged by ionic imbalance (Hazell, 2007).
Complexins and excitotoxicity:
Complexins are highly charged molecules found at the presynaptic nerve terminals and are key regulators of synaptic exocytosis (Fasshauer, 2008). They bind to the neuronal SNARE complex and are involved in calcium-dependent release of neurotransmitters. Mainly two forms, Complexin I (localised at the inhibitory synapses) and Complexin I,I (localised in the in axodendritic and axospinous synapses, which are mainly excitatory) are found in the human brain. It has been established that complexins are involved in the glutamate mediated excitotoxicity, but their clear roles are yet to be established. (Hazell, 2007)
Conclusion:
Thus it is quite clear that glutamate receptors play critical roles both directly and indirectly to mediate excitotoxicity following an ischemic stroke. The figure below shows a summary of the different roles played by glutamate receptors in triggering excitotoxicity due to stroke:
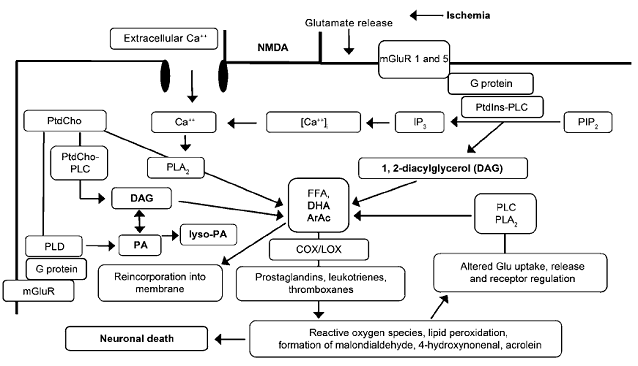
Fig. 6: A schematic representation of the biochemical cascades occurred during glutamate receptors mediated excitotoxicity as a consequence of stroke.
PLA 2, PtdCho-PLC, PtdIns-PLC, and PLD- phospholipases; DAG, PA, lyso-PA, DHA, and ArAc- lipid second messengers.
PLA 2 indicates phospholipase A 2 ; PtdCho-PLC: phosphatidylcholinephospholipase C; PtdIns-PLC: phosphatidylinositol-phospholipase C; PLD: phospholipase D; ArAc: arachidonic acid; DAG: 1,2-diacylglycerol; PA: phosphatidic acid; lyso-PA: lyso-phosphatidic acid; DHA: docosahexaenoic acid; COX/LOX: cyclooxygenases/lipoxygenases; PIP 2: phosphatidylinositol-4,5-bisphosphate; IP 3: inositol trisphosphate; mGluR: metabotropic glutamate receptor; NMDA: N-methyl-D-aspartate; FFA: free fatty acids; Glu: glutamate. [Adibhatla et al. (2006). The AAPS Journal. 8(2): E317]
There have been lots of research activities in recent times regarding the pathophysiology of excitotoxicity caused due to stroke. Researchers have been pretty successful in identifying the complex and overlapping pathways triggered due to excitotoxicity, which leads to ionic imbalance, oxidative and nitrosative stress, inflammation and apoptosis, by performing in vitro and in vivo experiments. The excitotoxic effects following a stroke are short-term, but the downstream effects are long lasting and so potentially life threatening.
Despite extensive basic research, the clinical trials for stroke-mediated excitotoxicity have been unsuccessful. Some of the proposed treatment strategies are employing NMDA-antagonists, antioxidants and enhancement of glutamate uptake. It is pretty clear that the intracellular Ca2+ is regulated by extracellular Ca2+ and Mg2+. So, controlling their concentrations will be beneficial in terms of blocking excitotoxicity (Lo, et al., 2003). Development of clot-lysing drugs is also a promising strategy in this field. More importantly, it would be better to prevent ischemic strokes rather than blocking excitotoxicity. Some of the risk factors for stroke are high blood pressure, high cholesterol level, diabetes, smoking; all these factors may lead to development of atherosclerosis (astherosclerosis) and thus blockade of blood vessels. Thus, leading an overall healthy life will be more beneficial, since it is very much true that ‘prevention is better than cure’.
References:
Adibhatla, R. M., Hatcher, J. F. and Dempsey, R. J. (2006). Lipids and Lipidomics in Brain Injury and Diseases. The AAPS Journal. 8: E314-E321
Bear, M. F., Connors, B. W. and Paradiso, M. A. (2007). Neuroscience: Exploring the Brain. 3rd ed. Philadelphia: Lippincott Williams and Wilkins.
Beilharz, E. J., Williams, C. E., Dragunow, M., Sirimanne, E. S. and Gluckman, P. D. (1995). Mechanisms of delayed cell death following hypoxic-ischemic injury in the immature rat: evidence for apoptosis during selective neuronal loss. Molecular Brain Research. 29: 1-14
Choi, D. W. (1987). Ionic Dependence of Glutamate Neurotoxicity. The Journal of Neuroscience. 7: 369-379
Dirnagl, U., Iadecola, C. and Moskowitz, M. A. (1999). Pathobiology of ischaemic stroke: an integrated view. Trends in Neurosciences. 22: 391-397
Fasshauer, D. (2008). Complexins [online]. Available from http://www.mpibpc.mpg.de/groups/jahn/Jahn/Complexin.htm [Accessed 16 April 2009].
Hazel, A. S. (2007). Excitotoxic mechanisms in stroke: An update of concepts and treatment strategies. Neurochemistry International. 50: 941-953
Ikeda, J., Terakawa, S., Murota, S. and Hirakawa, K. (1996). Nuclear Disintegration as a Leading Step of Glutamate Excitotoxicity in Brain Neurons. Journal of Neuroscience Research. 43: 613-622
Kristian, T. and Siesjo, B. K. (1998). Calcium in Ischemic Cell Death. Stroke. 29: 705-718
Lo, E. H., Dalkara, T. and Moskowitz, M. A. (2003). Mechanisms, Challenges and Opportunities in Stroke. Nature Reviews Neuroscience. 4: 399-415
MacDonald, J. F., Xiong, J.-G. and Jackson, M. F. (2006). Paradox of Ca2+ signaling, cell death and stroke. Trends in Neurosciences. 29: 75-81
Medical Research Council, Centre for Synaptic Plasticity and University of Bristol. (2009). Glutamate Receptors – Structure and Function [online]. Available from http://www.bris.ac.uk/Depts/Synaptic/info/glutamate.html [Accessed 16 April 2009].
Mody, I and MacDonald, J. F. (1995). NMDA receptor-dependent excitotoxicity: the role of Ca2+ release. Trends in Pharmaceutical Sciences. 16: 356-359
Moro, M. A., Almeida, A., Bolanos, J. P. and Lizasoain, I. (2005). Mitochondrial respiratory chain and free radical generation in stroke. Free Radical Biology & Medicine. 39: 1291 – 1304
Purves, D., Augustine, G. J., Fitzpatrick, D., Hall, W. C., LaMantia, A. S., McNamara, J. O. et al. (2008). Neuroscience. 4th ed. Massachusetts: Sinauer Associates Inc.
Rothman, S. M. and Olney, J. W. (1986). Glutamate and the Pathophysiology of Hypoxic-Ischemic Brain Damage. Annals of Neurology. 19: 105-111
Rothwell, N., Allan, S. and Toulmond, S. (1997). The Role of Interleukin 1 in Acute Neurodegeneration and Stroke: Pathophysiological and Therapeutic Implications. Jour. of Clin. Invest. 100: 2648-2652
Stanciu, M., Wang, Y., Kentor, R., Burke, N., Watkins, S., Kress, G., et al. (2000). Persistent Activation of ERK Contributes to Glutamate-induced Oxidative Toxicity in a Neuronal Cell Line and Primary Cortical Neuron Cultures. The Jour. Of Biol. Chem. 275: 12200-12206
Your Total Health. (2009). Stroke Center [online]. Available from http://yourtotalhealth.ivillage.com/stroke [Accessed 10 April 2009].