Surface-enhanced Raman scattering for therapeutic drug monitoring
Abstract
Therapeutic drug monitoring (TDM) involves measuring the serum level of a drug to ensure it is within the therapeutic window. It is essential for some drugs including the anticonvulsants such as phenytoin, carbamazepine and ethosuximide; the bronchodilator, theophylline and the tricyclic antidepressant, clomipramine. Current methods of analysis involve immunoassays and high performance liquid chromatography (HPLC), both of which are time consuming and require a dedicated laboratory. This paper investigates the potential use of surface enhanced Raman scattering (SERS) as a fast and efficient technique for TDM. Phenytoin exhibited a linear relationship within the therapeutic range of 4 x 10-5 – 8 x 10-5 M, with a lowest observable concentration of 5 x 10-7 M. Theophylline demonstrated a limited linear concentration range, with a lowest observable concentration of 2.5 x 10-4 M. This is slightly greater than the therapeutic window of 1.1 x 10-4 – 5.5 x 10-5 M. Carbamazepine and clomipramine both produced SERS spectra at a concentration of 1 x 10-3 M, but the poor signal-to-noise ratio prevented analysis at lower concentrations. In these cases, sample pre-concentration would be required. However, the selectivity of SERS is demonstrated as the spectra from each differ significantly, even though the molecular structure of carbamazepine and clomipramine is very similar. Ethosuximide produced no SERS spectrum when analysed using a range of metal surfaces with different aggregating agents. This suggests that derivatisation of this drug would be required in order to obtain efficient analyte adsorption for effective SERS. Analysis of the drugs in plasma resulted in high levels of fluorescence, necessitating the inclusion of a precipitation step to remove the plasma proteins if the technique was to be utilised for in situ drug monitoring.
1. Introduction
Therapeutic drug monitoring (TDM) is essential when patients are prescribed certain drugs. The purpose of TDM is to measure the serum level of a drug to ensure that the level is within the therapeutic range. TDM is useful for many reasons, particularly when there is poor correlation between the dosage and serum concentration for the drugs. If the drug is highly toxic, the therapeutic window may be small. Furthermore, when polypharmacy is used, certain drugs may alter the pharmacokinetics of the other drugs, resulting in a need for TDM1. It is particularly useful in special populations, such as the very young and very old, or patients with impaired renal or hepatic function2. There are many drugs that require TDM, including anticonvulsants, cardiac glycosides, theophylline, lithium, and tricyclic antidepressants3.
Phenytoin, ethosuximide and carbamazepine are all anticonvulsants used in the treatment of epilepsy. Carbamazepine is also used to treat trigeminal neuralgia and as a prophylaxis in lithium resistant cases of bipolar disorder. All these drugs have potential side effects such as leucopaenia, thrombocytopaenia, hepatitis and renal failure. Clomipramine is a tricyclic antidepressant of which the major side effect is cardiotoxicity. Theophylline is a bronchodilator used for reversible airway diseases such as asthma. Close monitoring of the theophylline serum concentration is necessary as the therapeutic window is extremely narrow, although some patients may have adverse effects within this range, with convulsions and arrhythmia being a sign of impending toxicity. Additionally, many factors can alter the metabolic rate of theophylline in the liver. For example, serum levels are increased in heart failure and cirrhosis but decreased in smokers and chronic alcoholics.
Currently, carbamazepine, phenytoin, ethosuximide and theophylline serum levels are all measured using immunoassays4. Although clomipramine can be measured in this way, the favoured method is high performance liquid chromatography (HPLC)5. These methods are adequate but are time consuming and require a dedicated laboratory. The time delay incurred can be frustrating for both doctor and patient. Therefore, an alternative analysis method, which could be carried out instantaneously on the ward or in general practitioners’ (GPs) surgeries, would be beneficial.
Surface enhanced Raman scattering (SERS) is a very sensitive and selective form of vibrational spectroscopy, allowing detection down to the single-molecule level6. SERS requires that the analyte be adsorbed onto, or in close proximity to, a suitably roughened metal surface, usually silver, gold or copper. Silver nanoparticles are the most commonly used SERS substrates, with the increase in Raman scattering efficiency arising from surface enhancement due to the interaction between the surface plasmons on the nanoparticles and the analyte molecule. This technique can provide an enhancement of the signal up to 106 over ordinary Raman scattering, bringing detection limits into the clinical range. SERS has several advantages compared to many other analytical methods. Standard Raman equipment, which is reliable, inexpensive and simple to operate, is used. Fluorescence, which is often a problem when ordinary Raman scattering is used, is quenched. Sharp, molecularly specific signals are obtained from aqueous solutions, allowing identification of a specific reagent in a sample and providing greater multiplexing ability than fluorescence. Additionally, no sample pre-treatment is required, resulting in much faster analysis times than with conventional methods such as HPLC. With the advent of microfluidics combined with hand-held portable spectrometers, the use of SERS as a detection system for clinical analysis is viable for use in GPs’ surgeries and in hospitals7.
SERS has been shown to be successful for the analysis of drugs of abuse, including amphetamine8-11, heroin, codeine and cocaine12. SERS has also been used for the analysis of clinical drugs, including 1,4-benzodiazepines drugs such as amphetamine and oxazepam13; danthron and quinizarin14; cisplatin, carboplatin and novel anti-cancer platinum complexes15; the antiretroviral drugs, hypercin and emodin16; beta blockers, propanolol, alprenolol, acebutolol and atenolol17; the diuretic drug, amiloride18; and the anti-cancer drug, doxorubicin19-21. Furthermore, quantitative analysis of the anticancer drug, mitoxantrone, has been reported using surface enhanced resonance Raman scattering (SERRS)22 and recently the analyses of clinical drugs using SERS with prior separation by HPLC was reported23. However, there is very little literature regarding the use of SERS for drugs which require TDM.
This paper investigates the potential use of SERS as an alternative method for the qualitative and quantitative analysis of a range of clinical drugs requiring TDM. A variety of modified silver and gold surfaces can be used but colloidal surfaces have been shown to provide sensitivity and to give a quantitative surface. Therefore, silver and gold colloids with various aggregating agents are investigated in order to obtain efficient analytic adsorption to the nanoparticles to provide an effective SERS.
2. Experiment
A silver colloid was prepared by the citrate reduction of silver nitrate, using the procedure reported by Lee and Meisel24, with conditions specified by Munro et al25. A gold colloid was prepared by the citrate reduction of sodium tetrachloroaurate using the method reported by Grabar et al26.
SERS spectra were recorded using a Renishaw system 200DR sandwich-scope equipped with a diode laser providing excitation at 532 nm. The sample was placed in the spectrometer and the laser focused in the middle of the sample. GRAMS software was used for data manipulation.
For use as aggregating agents, 1M solutions of sodium chloride in distilled water and 0.01 and 0.001 % solutions of poly-L-lysine hydrobromide (MWt 4000-15000) in distilled water were prepared.
Solutions of ethosuximide, clomipramine and theophylline were prepared in distilled water (1×10-2 M). Phenytoin and carbamazepine were prepared in acetone and methanol respectively (1×10-2 M). Further dilutions of the drugs were then carried out in the required solvents. Solutions of phenytoin and theophylline were also prepared in bovine serum.
500μl of colloid and 500μl of water were pipetted into a cuvette. 100μl of the analyte was added along with either 30μl of 0.001% poly-l-lysine, 50μl of 0.01% poly-L-lysine or 50μl of 1M NaCl. The cuvette was placed in the spectrometer and the samples were analysed for five accumulations of ten seconds each.
3. Results and Discussion
Two essential conditions are required to obtain effective SERS; the analyte must be adsorbed on the roughened metal surface and the plasmon resonance frequency of the metal must be close to that of the excitation frequency used. Both gold and silver colloids are negatively charged, thus only molecules which are positively charged or have strong chemisorption will give effective SERS. By aggregating the colloid, the surface plasmon can be shifted to match the excitation frequency. However, different aggregating agents will active aggregation by different mechanisms, resulting in different surface chemistry on the metal surface. Here, we compare the most widely used aggregating agent, sodium chloride, with the aggregating agent poly-L-lysine which has been shown to be effective, but for which there is limited data.
The molecular structures of phenytoin, theophylline, carbamazepine, clomipramine and ethosuximide are shown in Figure 1. Each drug was analysed using silver and gold colloid aggregated with 0.001% poly-L-lysine, 0.01% poly-L-lysine or 1M NaCl. Table 1 shows the optimum conditions for SERS analysis of each drug. None of the drugs produced a SERS spectrum when gold colloid was used.
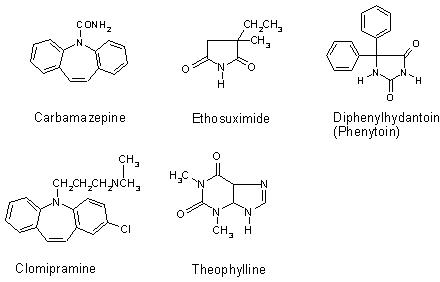
Figure 1. Molecular structures of the clinical drugs analysed.
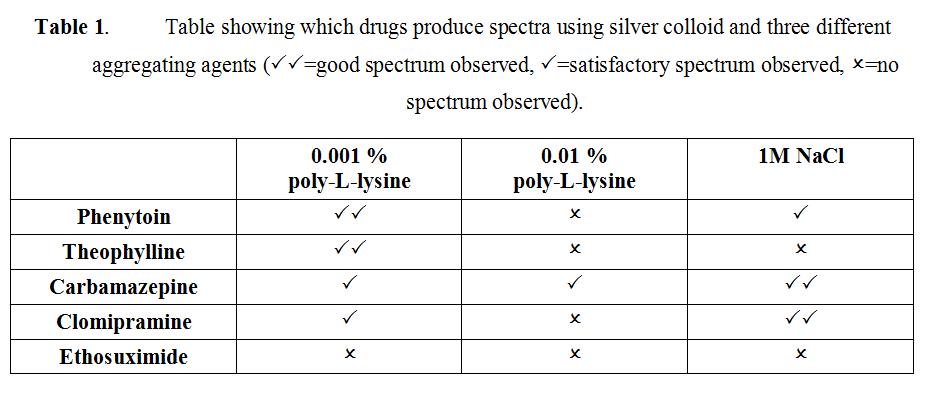
Figure 1. Molecular structures of the clinical drugs analysed.
Phenytoin and theophylline produced SERS with good signal-to-noise ratios over a wide concentration range when aggregated with 0.001 % poly-L-lysine (Figure 2). Under these conditions, a concentration study of phenytoin exhibited a linear relationship over the whole range of concentrations analysed (Figure 3), with a lowest detectable concentration of approximately 5 x 10-7 M. The optimum plasma concentration of phenytoin is between 4 x 10-5 and 8 x 10-5 M. Therefore, the concentration range detectable is within the therapeutic window. Theophylline exhibited a linear relationship over a limited concentration range when aggregated with 0.001 % poly-L-lysine, (Figure 4), with a lowest detectable concentration of approximately 2.5 x 10-4 M. To achieve adequate bronchodilation, the plasma concentration of theophylline needed is between 1.1 x 10-4 and 5.5 x 10-5 M, although adequate effects may be encountered at levels lower than this. If the dilution of the drug with the colloid, water and aggregating agent is taken into account, the lowest detectable concentration equates to 2.21 x 10-5 M. Thus, the detection limit of theophylline is just within the clinical range. Therefore, SERS is a suitable detection method for assay development to determine plasma concentration of both drugs. Further improvements in sensitivity are possible by integrating a flow-cell set-up into the technique by means of a microfluidics chip. Presently, due to the large sample size (1130 l) only a small volume is actually probed by the laser. With a microfluidics chip, a much smaller volume of sample is required, but the percentage of the sample exposed to the laser beam is much greater, resulting in greater sensitivity. If this is combined with a portable spectrometer, potentially, a hand-held device could be manufactured7. This would allow TDM to be performed either on hospital wards or in GPs’ surgeries, with results being available instantly.
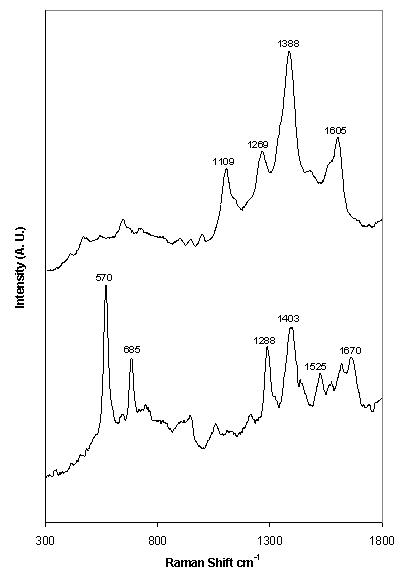
Figure 2. SERS spectrum of (top) 1 x 10-4 M phenytoin and (foot) 1 x 10-2 M theophylline
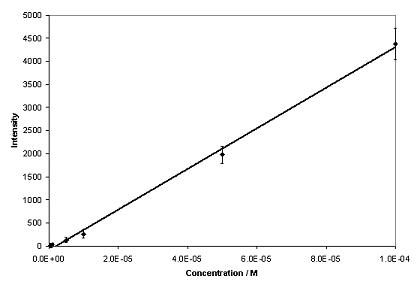
aggregated with 0.001% poly-L-lysine. Excitation at 532nm. Figure 3. Calibration graph for phenytoin (n=5).
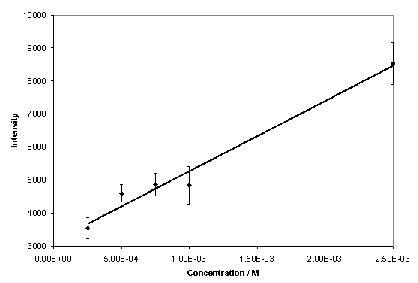
Figure 4. Calibration graph for theophylline over a limited concentration range (n=5).
Carbamazepine and clomipramine give good examples of the molecular specificity of SERS. The molecular structure of both drugs is very similar (Figure 1); however, the spectra produced differ significantly (Figure 5), allowing specific drugs to be recognised in situ. Carbamazepine produced a good SERS spectrum at 1 x 10-3 M when aggregated with 1 M NaCl. However, no peaks were observed at any other concentrations. Clomipramine also produced a spectrum at 1 x 10-3 M, but the poor signal-to-noise ratio prevented analysis at lower concentrations. Thus, the use of SERS for TDM of carbamazepine and clomipramine shows great potential, but, in order for SERS to be used as a quantitative analytical technique for these drugs, an improvement in the sensitivity is required. This may be obtained without prior derivatisation or separation of the drug with the use of a flow-cell system. Alternatively, efficient analyte adsorption to the metal surface using different aggregating agents may be effective, as would pre-concentration of the sample using a column. Additionally, derivatisation of the drug to incorporate a silver surface-seeking group, such as benzotriazole, would ensure adequate adsorption of the drug to the surface27. The inclusion of a chromophore would allow resonance conditions to be used, resulting in additional enhancement. This would also help expand SERS, making it a universal, economically viable technique for a wide variety of drugs. An example of this is ethosuximide, which failed to produce detectable SERS under the conditions used here.
Table 1 highlights the importance of the correct choice of SERS substrate and aggregating agents to ensure that effective SERS spectra are obtained. In particular, the substrate surface onto which the analyte is adsorbed plays a crucial role in determining whether SERS spectra are obtained. Phenytoin and theophylline produced the best spectra when aggregated with 0.001% poly-L-lysine on silver nanoparticles. In comparison, clomipramine and carbamazepine gave the most effective SERS when aggregated with 1M NaCl and adsorbed onto a silver surface. Silver and gold colloid both have negatively charged surfaces due to the stabilising citrate layer25. When sodium chloride is added, silver chloride is formed and the surface stays negatively charged. However, poly-L-lysine is an organic aggregating agent with a net positive charge. If a small amount of poly-L-lysine is added (10 μl 0.001 %), the colloid remains negatively charged, but upon further titration, the surface becomes positively charged. A recent study has found that the optimum amount of poly-L-lysine required to form a positively charged colloid is 30μl of 0.001%28. An initial evaluation of the results would suggest that carbamazepine and clomipramine are adsorbed when the surface of the silver colloid remains negatively charged. Conversely, phenytoin and theophylline both produce the best spectra when the silver colloid has been modified to have a net positive charge. These parameters need to be investigated further to optimise the SERS enhancement from carbamazepine and clomipramine and to allow SERS to be obtained from a variety of other drugs with different molecular structures.
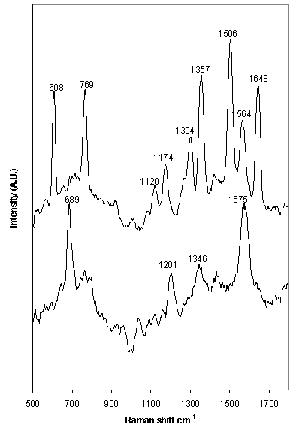
Figure 5. SERS spectrum of (top) 1 x 10-3 M carbamazepine and (foot) 1 x 10-3 M clomipramine aggregated with 1 M NaCl. Excitation at 532nm.
Phenytoin and theophylline were both analysed in bovine serum to mimic a human sample. Serum often presents a problem in analyses using Raman scattering as it fluoresces strongly, resulting in the need for prior separation of the drug from the serum by means of centrifugation. SERS offers a unique answer to this problem as fluoresce is quenched when the analyte molecule is in close proximity to the metal surface, providing an alternative, non-radiative route for energy loss29,30. This has been proven with mitoxantrone in a flow-system, where the drug adsorbs to the silver nanoparticles, producing a very strong SERRS. Dilution of the serum by the flowing colloid stream also aids a reduction in fluorescence22. However, a large background fluorescence was observed when theophylline and phenytoin were analysed in serum, suggesting the drug does not adsorb strongly to the silver colloid. Simple derivatisation of the drugs to incorporate a surface-seeking group may overcome this problem to ensure more efficient analyte adsorption to the colloid. Fluorescence may also be reduced if a flow-cell was used, as serum dilution would occur. Alternatively, a single chromatography step may be incorporated to remove the plasma proteins. This would have the additional benefit of concentrating the drug, thereby increasing the sensitivity of SERS. This is an important area of further research if SERS is to be used for TDM.
References
1. A. E. Balant-Gorgia, M. Gex-fabey and L. P. Balant, Therapie, 1996, 51, 399.
2. P. D. Walson, Clin. Chem., 1998, 44, 415.
3. S. H. Preskorn, R. C. Dorey and G. S. Jerkovich, Clin. Chem., 1988, 34, 822.
4. E. L. Frank, E. L. Schwarz, J. Juenke, T. H. Annesley and W. L. Roberts, Am. J. Clin. Pathol., 2002, 118, 124.
5. H. Weigmann, S. Hartter and C. Hiemke, J. Chromatogr. B Biomed. Sci. Appl., 1998, 710, 227.
6. K. Kneipp, Y. Wang and H. Kneipp, Phys. Rev. Lett.. 1997, 78, 1667.
7. F. T. Docherty, P. B. Monaghan, R. Keir, D. Graham, W. E. Smith and J. M. Cooper, Chem. Commun., 2004, 118.
8. B. Saegmuller, G. Brehm and S. Schneider, Appl. Spectrosc., 2000, 54, 1849.
9. B. Sagmuller, B. Schwarze, G. Brehm and S. Schneider, Analyst, 2001, 126, 2066.
10. R. A. Sulk, R. C. Corcoran and K. T. Carron, Appl. Spectrosc., 1999, 53, 954.
11. K. Faulds, W. E. Smith, D. Graham and R. J. Lacey, Analyst, 2002, 127, 282.
12. E. Horvath, J. Mink and J. Kristof, Mikrochim. Acta [suppl.], 1997, 14, 745.
13. G. Trachta, B. Schwarze, G. Brehm, S. Schneider, M. Hennemann and T. Clark, J. Raman Spectrosc., 2004, 35, 368.
14. G. Fabriciova, S. Sanchez-Cortes, J. V. Garcia-Ramos and P. Miskovsky, J. Raman Spectrosc., 2004, 35, 384.
15. B. Giese, G. B. Deacon, J. Kuduk-Jaworska and D. McNaughton, Biopolymers, 2002, 67, 294.
16. S. Sanchez-Cortes, D. Jancura, P. Miskovsky and A. Bertoluzza, Spectrochim. Acta A, 1997, 53, 769.
17. A. Ruperez and J. J. Laserna, Anal. Chim. Acta, 1996, 335, 87.
18. N. Calvo, R. Montes and J. J. Laserna, Anal. Chim. Acta, 1993, 280, 263.
19. A. Lorén, C. Eliasson, M. Josefson, K. V. G. K. Murty, M. Käll, J. Abrahamsson and K. Abrahamsson, J. Raman Spectrosc., 2001, 32, 971.
20. A. Beljebbar, G. D. Sockalingum, J. F. Angiboust and M. Manfait, Spectrochim. Acta A, 1995, 51, 2083.
21. C. Eliasson, A. Lorén, K. V. G. K. Murty, M. Josefson, M. Käll, J. Abrahamsson and K. Abrahamsson, Spectrochim. Acta A, 2001, 57, 1907.
22. C. McLaughlin, D. MacMillan, C. McCardle and W. E. Smith, Anal. Chem., 2002, 74, 3160.
23. G. Trachta, B. Schwarz, B. Sagmuller, G. Brehm and S. Schneider, J. Mol. Struct., 2004, 694, 175.
24. P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391.
25. C. H. Munro, W. E. Smith, M. Garner, J. Clarkson and P. C. White, Langmuir, 1995, 11, 3712.
26. K. C. Grabar, R. G. Freeman, M. B. Hommer and M. J. Natan, Anal. Chem., 1995, 67, 735.
27. G. McAnally, C. McLaughlin, R. Brown, D. C. Robson, K. Faulds, D. R. Tackley, W. E. Smith and D. Graham, Analyst, 2002, 127, 838.
28. R. E. Littleford, Unpublished work, University of Strathclyde, 2004.
29. O. Siiman, A. Lepp and M. Kerker, J. Phys. Chem., 1983, 87, 5319.
30. E. J. Zeeman, K. T. Carron and G. C. Shatz, J. Chem. Phys., 1987, 87, 4189.