Iodine in the Air: origin, transformation and exchange to mammals
Abstract
As part of its bio-geochemical cycle, the injection of iodine-containing gases into the atmosphere and their subsequent chemical transformation therein, play a crucial role in environmental and health aspects associated with iodine – most importantly, in determining the quantity of the element available to the mammalian diet. This review focuses on these processes and the variety of gas- and aerosol-phase species which constitute the terrestrial iodine cycle, through discussion of the origin and measurement of atmospheric iodine in its various forms (section 2). Potential health and environmental issues connected with atmospheric iodine are also reviewed (section 3), along with discussion of the consequences of the release of radioactive iodine (I-131) into the air from nuclear reactor accidents and weapon tests which have occurred over the past half-century or so (section 4).
1. Introduction
Iodine is an essential trace element in the endocrine system, necessary for the production of the hormones triiodothyronine (T3) and thyroxine (T4) in the thyroid gland. Mammals thus provide the termination step for the cycling of iodine in the biosphere. This biogeochemical cycle (Figure 1) involves processes of oceanic release, sea-air transfer, photo-chemical transformation, aerosol uptake and deposition to land where iodine is adsorbed onto soil and vegetation (Fuge and Johnson, 1986).
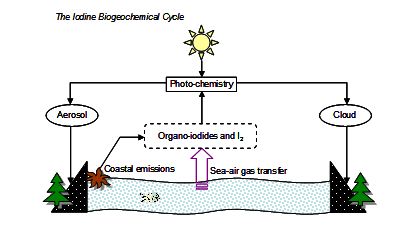
Figure 1 Schematic of the production, transfer and loss processes for atmospheric iodine.
The primary source of iodine is marine fauna and flora, with microalgae (phytoplankton and cyanobacteria) in open oceans and macroalgae (seaweed) in coastal areas releasing a range of gas-phase iodine-containing organic species including methyl iodide (CH3I) and diiodomethane (CH2I2), and molecular iodine (I2) to the atmosphere (Vogt, 1999; Carpenter, 2003). The predominant loss process of these molecules is photolysis, with subsequent oxidation by ozone (O3) and reaction with other atmospheric species (e.g. OH and NO2) in the marine boundary layer (MBL) leading to the formation of iodine sinks in the form of more stable gas-phase species and aerosol particles (Vogt et al., 1999; McFiggans et al., 2000; O’Dowd and Hoffmann, 2005). There is therefore an intricate interplay between physical and chemical atmospheric processes prior to the eventual deposition of iodine to soil and ultimately ingestion by animals and humans.
2. Sources and Measurements of Atmospheric Iodine
Despite the first detailed measurements of air-borne iodine dating back to the 1930’s (Cauer, 1936, 1939), and observations of iodine enrichment (with respect to chlorine) in rainfall and aerosol (compared with seawater) in the 1960’s (Duce et al., 1963), it was only a decade later, with the direct measurement of CH3I in Atlantic water and air (Lovelock et al., 1973) that details of the origin and speciation of atmospheric iodine began to be resolved. Since then, and particularly in the last decade, research in this area has exploded and the role and potential climatic effects of iodine in the atmosphere have become to be put into more accurate context (Kolb, 2002; von Glasow, 2005). A current estimate of the total emission rate of organo-iodide gases into the atmosphere lies in the range 2-4.5 Tg (1012 g) per year (Saiz-Lopez and Plane, 2004a), with CH3I and CH2I2 accounting for > 90% of this atmospheric input. For comparison, the estimated input from the major anthropogenic source of iodine (fossil fuel burning) is some 2-3 orders of magnitude smaller (Vogt, 1999).
A number of organo-iodide species are of biological origin (Gribble, 2003) and have been identified at different locations around the globe – see Vogt (1999) or Carpenter (2003) for a summary of some of these more recent measurements. Measured atmospheric volume mixing ratios of CH3I for example, show an order of magnitude increase at seaweed-rich coastal locations compared with typical oceanic air values of 1-3 parts per trillion or ppt (1 ppt = 2.5 × 107 molecules cm-3). Only very recently, molecular iodine (I2) was measured in the MBL for the first time, at Mace Head, Ireland (53°N), with peak mixing ratios of 80-90 ppt at evening low-tide periods (Saiz-Lopez and Plane, 2004b) – see Figure 3. These last two observations along with reported correlations between measured gas-phase iodine oxide (IO and OIO) levels and rapid aerosol particle nucleation events or ‘bursts’ (O’Dowd and Hoffmann, 2005) have established that the most probable mechanism for the release of these iodine-containing gases into the marine air is initiated by a stress-induced biochemical pathway. For example, at Mace Head the coastline is rich in the brown kelp, Oarweed (Laminaria Digitata). These accumulate very high concentrations of iodine within their structure (Leblanc et al., 2006) and recent laboratory studies have shown that light-, chemical- and oxidative-induced stress in such seaweed species result in the release of gases including CH2I2 and I2 (Palmer et al., 2005). Saiz-Lopez and Plane (2004b) estimated an annual flux for I2 of coastal origin, of 0.5 Tg, assuming that 20% of the Earth’s total coastline (1.6 × 106 km) is suitably habitable for these iodine-rich seaweed types.
In terms of the open ocean, whilst algal sources are most likely for the organo-iodides, a number of alternative processes have been suggested for volatilisation of I2 from seawater to the air. These include (i) the reduction of iodide ions (I–) by UV photo-oxidation in the presence of O2, originally proposed by Cauer (1939) and verified experimentally by Miyake and Tsunogai (1963), (ii) oxidation of I– by O3 (Garland and Curtis, 1981) and (iii) decomposition of iodine-rich organics at the ocean surface and on the surface of sea salt particles generated and passed into the air via wind-driven wave breaking (Moyers and Duce, 1972; Seto and Duce, 1972). Although estimates were made in the cited studies for likely global annual production rates of iodine from such mechanisms ((i) 0.4 Tg per year, (ii) 0.1 Tg per year), no concerted effort has been undertaken to more accurately characterise these processes and quantify the resultant gas emissions. Consequently, even though these sources are potentially far greater than the localised coastal emissions due to the large sea surface area available, the contribution of the open ocean to the atmospheric iodine burden remains an unresolved issue.
3. Health and Environment Impacts
In the 1960’s, a series of studies reported the beneficial role of iodine in the atmosphere with regard to issues such as the inhibition of urban photo-chemical smog production and artificial weather modification. Stephens et al. (1962) showed that addition of elemental iodine dramatically suppressed the formation of organic products such as aldehydes and peroxyacetyl nitrate (PAN) produced from the photo-chemical reactions between alkenes and NO2 but concluded that the high concentrations of iodine required (parts per million, ppm) precluded any large-scale practical application for its use in smog reduction. Soon after, Hamilton et al. (1963) reported the effective inhibition of O3 formation by trace amounts of iodine in a controlled smog-filled environment containing a mixture of car exhaust fumes and NO2 and the resulting health benefits observed in test subjects exposed to the chemical pollutants. This method was suggested as a practical solution to the removal of ozone in supersonic aircraft and indeed the authors subsequently took out a patent (#3,084,024 – US Patent Office) on behalf of the Lockheed Aircraft Corporation.
Schaefer (1966) reported the activation of large numbers of ice nuclei upon the addition of trace levels of iodine vapour to car exhaust (containing lead oxide nanoparticles) at temperatures from -3 to -20°C in the laboratory. The formation of lead iodide was concluded to have a similar ‘seeding’ effect to silver iodide particles (Vonnegut, 1947), which had been used in an attempt to artificially modify cloud properties and enhance precipitation. Consequently this method was proposed as a means of the removal of harmful aerosol formed in polluted urban areas and also in artificial weather modification. However, the development of unleaded fuels, for which no similar ice nucleating ability was shown to occur in the presence of iodine (Hogan, 1967), provided a better long-term solution to this problem.
Vikis and MacFarlane (1985) reported on reaction rates between I2 and O3 and the resultant formation of solid-phase iodine oxide aerosol. Coming from the opposite direction to the earlier work of Hamilton et al. (1962), this led the authors to suggest that addition of O3 to nuclear reactor environments should be considered as a practical route for the removal of air-borne radioactive iodine species produced as fission by-products (see next section).
Almost certainly however, the most important impact of the presence of iodine in the atmosphere is its potential for depletion of O3 in the troposphere and lower stratosphere. Results of laboratory studies of the photo-oxidation of I2 (Jenkin et al., 1985) supported the notion that oxidation of iodine released by photolysis of precursor gases would lead to a catalytic destruction cycle with the net result that 2 molecules of O3 are converted to 3 molecules of O2. Davis et al. (1996) predicted that, for equatorial to mid-latitude regions (0-42°), a total gas-phase iodine mixing ratio (ΣI) of just 1.5 ppt in the upper troposphere would result in a 6% depletion of ozone, increasing to 30% for ΣI = 7 ppt. It should be noted that their calculations did not include an I2 source from the oceans in addition to the organo-iodides. A decade later, with improved knowledge of reaction pathways and rate constants, a study by Saiz-Lopez et al. (2006b) of coastal, daytime I2 emissions (average value of 6.5 ppt) at Mace Head, Ireland predicted an O3 loss rate in the MBL, along a 4.2 km path-length from the coast, of 1 ppb (10-9) per hour, corresponding to an hourly depletion of ~ 4%. Modelling of iodine chemistry under a polar scenario (Barrow, Alaska 71°N), which included I2 as a source, also predicted a significant O3 depletion contribution in the spring-time (Calvert and Lindberg, 2004b).
Solomon et al. (1994) implicated iodine in lower stratospheric ozone loss as a result of the rapid vertical transport of precursor gases via convection currents resulting in photolysis and subsequent chemical transformations at altitudes up to 20 km and concluded that this route would be of the order of 3 orders of magnitude greater than O3 loss resulting from chlorine chemistry.
4. Radioactive Iodine – atmospheric sources and consequences
The stable I-127 isotope constitutes 100% of naturally occurring iodine. However a number of artificial, radioactive isotopes are formed as by-products of nuclear fission pathways (see Figure 2). Of these, I-131 is the most insidious in the biogeochemical cycle (via its transport in the atmosphere) due to its subsequent assimilation in the thyroid gland and relatively short half-life of ~ 8 days.
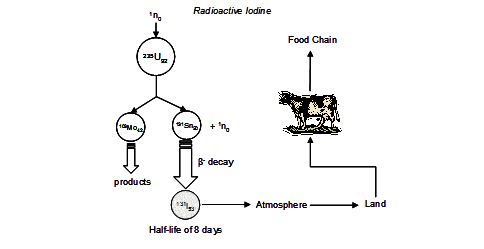
Figure 2 Nuclear fission route to the formation of radioactive I-131 and subsequent pathways to its ingestion.
Once released into the air, wind dispersal followed by deposition to soil via rainfall and aerosol uptake and adsorption onto vegetation (Chamberlain et al. 1960) leads to the primary source of I-131 in humans originating from milk produced by grazing animals. Infants are therefore especially susceptible to accumulation of this isotope to levels at which carcinoma can occur.
Jenkin et al. (1985) estimated that if radioactive iodine is released primarily as I2, then during daytime, 95% will be photolysed within 1 minute of release and so deposition during daylight hours is likely to occur in the form of species such as IONO2 or aerosol whereas at night-time, the major deposition channel is more likely to be in the elemental form.
The major atmospheric source of I-131 originates from nuclear reactors in which the radioactive by-products are normally contained by the reactor coolant. However, since the first electricity-generating power plant went on-line in 1954, a handful of accidents, resulting in the release of radio-isotopes to the air, have occurred and the subsequent instances of contaminated milk and increased thyroid cancer cases have been well documented. For example, the accident at the Windscale reactor in Cumbria, England in 1957, is estimated to have released 2 × 104 Curies (Ci) of I-131 into the atmosphere (Williams, 2006), equivalent to ~ 7.5 × 1014 disintegrations per second. Approximately a year after the accident, measurements of different radioactive isotopes collected in air filters and from grass samples showed that I-131 had by far, the highest activity (Chamberlain and Dunster, 1958). Wind dispersal carried the fallout along the coastline and further inland i.e. to Leeds, Yorkshire, where subsequent studies indicated elevated I-131 concentrations in milk and in infant thyroids (Burch, 1959).
However, this incident was dwarfed in scale by the meltdown which took place at the Chernobyl reactor in the former Soviet Union or modern day Ukraine in 1986. Setting Windscale at 1 on an I-131 release scale, Chernobyl comes in at ~ 2300 (Williams, 2006) and within a few years, reports were made of a marked increase in cases of thyroid cancer in neighbouring areas such as modern day Belarus (Kazakov et al., 1992). Studies of the aftermath of the incident continue today although much uncertainty remains as to the long-term environmental and health effects (Baverstock and Williams, 2006). Still greater than even this event, in terms of radioactive iodine release, was the cumulative release (Windscale ×7500) from the atomic weapon test program conducted in Nevada, USA from 1951-1962. Again, fallout of the isotope was directly linked with radioactivity detected in cattle (Van Middlesworth, 1956) and in later studies, air trajectory calculations of fallout from underground explosions showed how effectively and rapidly, air-borne I-131 could be transported over large distances prior to deposition (Martell, 1964).
In conclusion, such relatively short term studies only hint at the likely scale of effects possible following such incidents if fast, remedial action is not taken, and harmful levels of I-131 are transported through the atmosphere prior to deposition back to land and incorporation into the food chain. No further major reactor accidents have taken place since Chernobyl and nuclear weapon tests are much less common than 40-50 years ago. However in the light of the current build-up of such weapons in places such as North Korea, Pakistan and the Middle East, and serious consideration, at least in the UK, of recommencing a program of nuclear reactor construction as a means of combating reliance on fossil fuel burning, lessons from the recent past must be kept in mind.
5. Summary
- The main sources of atmospheric iodine are biogenic, i.e. phytoplankton in the open ocean and certain seaweed species at coastal sites, with some likely contribution from chemical transformation of I– in seawater.
- These marine species release organic iodine gases (i.e. CH3I and CH2I2) and molecular iodine (I2).
- The total flux of these iodine-containing-gases into the air from the oceans is ~ 3.0 – 5.5 × 1012 g per year.
- The major impact of atmospheric iodine chemistry is the resultant depletion of O3, while other consequences, such as enhanced cloud formation, remain to be established.
- The release of radioactive iodine isotopes, particularly I-131, into the atmosphere and their subsequent transport and deposition, has conclusively been linked to increases in cases of infant thyroid cancer.
References
Baker, A.R., Tunnicliffe, C. and Jickells, T.D. (2001). J. Geophys. Res. 106, 28743-28749.
Baker, A.R. (2005). Environ. Chem. 2, 295-298.
Baverstock, K. and Williams, D. (2006). Environ. Health Perspect. 114, 1312-1317.
Brooks, S.B., Saiz-Lopez, A., Skov, H., Lindberg, S.E., Plane, J.M.C., and Goodsite, M.E. (2006). Geophys. Res. Lett. 33, L13812, pp. 1-4.
Burch, P.R.J. (1959). Nature 183, 515-517
Calvert, J.G., and Lindberg, S.E. (2004a). Atmos. Env. 38, 5105-5116.
Calvert, J.G., and Lindberg, S.E. (2004b). Atmos. Env. 38, 5087-5104.
Carpenter, L.J. (2003). Chem. Rev. 103, 4953-4962.
Cauer, H. (1936). Fresenius’ J. Anal. Chem. 104, 161-169.
Cauer, H. (1939). Angew. Chem. 52, 625-628.
Chamberlain, A.C. and Dunster, H.J. (1958). Nature 182, 629-630.
Chamberlain, A.C., Eggleton, A.E.J., Megaw, W.J. and Morris, J.B. (1960). Disc. Farad. Soc. 30, 162-169.
Chatfield, R.B., and Crutzen, P.J. (1990). J. Geophys. Res. 95, 22319-22341.
Davis, D., Crawford, J., Liu,S., McKeen , S., Bandy, A., Thornton, D., Rowland, F. and Blake, D. (1986). J. Geophys. Res. 101, 2135-2147.
Duce, R.A., Wasson, J.T., Winchester, J.W. and Burns, F. (1963). J. Geophys. Res. 68, 3943-3947.
Fuge, R. and Johnson, C.C. (1986). Environ. Geochem. Health 8, 31-54.
Garland, J.A. and Curtis, H. (1981). J. Geophys. Res. 86, 3183-3186.
Gravestock, T., Blitz, M.A., and Heard, D.E. (2005). Phys. Chem. Chem. Phys. 7, 2173-2181.
Gribble, G.W. (2003). Chemosphere 52, 289-297.
Hamilton, W.F., Levine, M. and Simon, E. (1963). Science 140, 190-191.
Hogan, A.W. (1967). Science 158, 800.
Jenkin, M.E., Cox, R.A. and Candeland, D.E. (1985). J. Atmos. Chem. 2, 359-375.
Jimenez, J.L., et al., (+8 co-authors) (2003). J. Geophys. Res. 108, 4318, 1-25.
Kazakov, V.S., Demidchik, E.P. and Astakhova, L.N. (1992). Nature 359, 21.
Kolb, C.E. (2002). Nature 417, 597-598.
Leblanc, C., et al. (+13 co-authors) (2006). Biochimie 88, 1773-1785.
Lindberg, S.E., and Stratton, W.J. (1998). Environ. Sci. Technol. 32, 49-57.
Lovelock, J.E., Maggs, R.J. and Wade, R.J. (1973). Nature 241, 195-196.
Martell, E.A. (1964). Science 143, 127-129.
McFiggans, G., Plane, J.M.C., Allan, B.J., Carpenter, L.J., Coe, H. and O’Dowd, C.D. (2000). J. Geophys. Res. 105, 14371-14385.
McFiggans, G., et al. (+12 co-authors) (2004). Atmos. Chem. Phys. 4, 701-713.
Miyake, S. and Tsunogai, S. (1963). J. Geophys. Res. 68, 3989-3994.
Moyers, J.L. and Duce, R.A. (1972). J. Geophys. Res. 77, 5229-5238.
O’Dowd, C.D. and Hoffmann, T. (2005). Environ. Chem. 2, 245-255.
Palmer, C.J., Anders, T.L., Carpenter, L.J., Küpper, F.C. and McFiggans, G. (2005). Environ. Chem. 2, 282-290.
Saiz-Lopez, A., Saunders, R.W., Joseph, D.M., Ashworth, S.H. and Plane, J.M.C. (2004). Atmos. Chem. Phys. 4, 1443-1450.
Saiz-Lopez, A. and Plane, J.M.C. (2004a). J. Phys. IV 121, 223-238.
Saiz-Lopez, A. and Plane, J.M.C. (2004b). Geophys. Res. Lett. 31, L04112, pp. 1-4.
Saiz-Lopez, A., Shillito, J.A., Coe, H., and Plane, J.M.C. (2006a). Atmos. Chem. Phys. 6, 1513-1528.
Saiz-Lopez, A., Plane, J.M.C., McFiggans, G., Williams, P.I., Ball, S.M., Bitter, M., Jones, R.L., Hongwei, C. and Hoffmann, T. (2006b). Atmos. Chem. Phys. 6, 883-895.
Saunders, R.W. and Plane, J.M.C. (2005). Environ. Chem. 2, 299-303.
Saunders, R.W. and Plane, J.M.C. (2006). J. Aero. Sci. 37, 1737-1749.
Seto, F.Y.B. and Duce, R.A. (1972). J. Geophys. Res. 77, 5339-5349.
Solomon, S., Garcia, R.R. and Ravishankara, A.R. (1994). J. Geophys. Res. 99, 20491-20499.
Stephens, E.R., Linnell, R.H. and Reckner, L. (1962) Science 138, 231-832.
Van Middlesworth, L. (1956). Science 123, 982-983.
Vikis, A.C. and R. MacFarlane (1985) J. Chem. Phys. 89, 812-815.
Vogt, R. (1999). In ‘Reactive Halogen Compounds in the Atmosphere’, (Volume 4E of ‘The Handbook of Environmental Chemistry’) pp. 113-128, Springer, Berlin.
Vogt, R., Sander, S., von Glasow, R. and Crutzen, P.J. (1999). J. Atmos. Chem. 32, 375-395.
von Glasow, R. (2005). Environ. Chem. 2, 243-244.
Vonnegut, B. (1947). J. Appl. Phys. 18, 593-595.
Williams, E.D. (2006). J. Surg. Oncol. 94, 670-677.